
PREGUNTAS
Antes de continuar intente contestar las siguientes preguntas. Las respuestas podrán ser encontradas al final del artículo, junto a una explicación.
- Los opioides producen analgesia activando principalmente (mejor respuesta única)
a. Receptor Mu (MOP)
b. Receptor delta (DOP)
c. Receptor kappa
d. Receptor de orfanina/nociceptina - El efecto analgésico pico de la morfina ocurre en (mejor respuesta única)
a. 1-2 min
b. 5-10 min
c. 30-60 min
d. 1-2 hs - Los receptores opioides (verdadero o falso)
a. Son proteínas G acopladas
b. Activan sistemas de segundos mensajeros intracelulares
c. Muestran agonismo parcial
d. Nunca se internalizan
e. Son encontradas en células inmunes
INTRODUCCIÓN
Los analgésicos opioides son el gold standard (patrón oro) en analgesia sistémica para el dolor severo agudo. Hay muchos compuestos diferentes en uso clínico en todo el mundo. Coste, regulaciones y ajustes clínicos dictan su disponibilidad. Hay una variabilidad intra-paciente y entre los pacientes en la respuesta a los opioides. La tolerancia cruzada incompleta ocurre cuando un paciente se cambia de un opioide a otro, la implicación clínica es que las dosis de opioides equivalentes deberán reducirse al comenzar un nuevo opioide para evitar sobredosis. El conocimiento de las diferencias farmacológicas entre los opioides puede aplicarse para seleccionar el fármaco adecuado para el entorno clínico relevante y minimizar el impacto de los efectos secundarios. En los últimos 20 años ha salido a la luz, la mayor información acerca de la farmacodinamia y la farmacocinética en términos de dímeros y oligómeros de receptores opioides, efectos del sistema de segundos mensajeros y el genotipado.
Los analgésicos opioides ejercen sus acciones farmacológicas a través del receptor μ-opioide, MOP, y algunas además poseen actividad del κ-receptor opiáceo, KOP.
RECEPTORES OPIOIDES
El receptor opioide, es una proteína G acoplada, con siete regiones transmembrana. Está actualmente clasificado en:
- DOP o receptor delta δ (llamado así por el tejido donde fue aislado por primera vez en el conducto deferente)
- KOP o receptor kappa κ ( llamado así, por su primer ligando, Ketociclazina)
- MOP o receptor mu μ ( llamado así por la Morfina, propuesto en 1976, y clonado en 1993)
Los receptores, fueron temporariamente renombrados en 1996 por la Unión Internacional de Farmacología (UIFar) como OP1, OP2 y OP3. Anterior a esto, eran conocidos como DOR, KOR y MOR. Debido a la extensa literatura usada en la nomenclatura griega, la UIFar ha recomendado el uso de δ ,κ y, μ intercambiable con DOP, KOP, y MOP.
Localización
Esos receptores están ubicados dentro del sistema nervioso central, en el cerebro medio, áreas del tronco cerebral asociadas con las vías moduladoras descendentes y en el cuerno dorsal de la médula espinal. También hay sitios periféricos, incluyendo los conductos deferentes, articulación de la rodilla, el tracto gastrointestinal, el corazón y el sistema inmunológico.
Analgesia opioide
La activación de los receptores opioides del cerebro medio, indirectamente, estimulan las vías descendentes inhibitorias. Estas vías descendentes involucran la transmisión serotoninérgica y noradrenérgica que resulta en la inhibición del tráfico nociceptivo en la sustancia gelatinosa del asta dorsal de la médula espinal. Además los opioides pueden actuar directamente sobre las neuronas nociceptivas en el asta dorsal y la periferia.
Receptor de Nociceptina
Este receptor conocido como NOP, fue descubierto en 1984. Su ligando endógeno es la nociceptina /orfanina (FQ). A diferencia de los receptores de opioides clásicos, no se liga a naloxona, lo que ha llevado a la sugerencia de que no debe clasificarse como parte de la familia de receptores de opioides. Sin embargo, tiene una estructura y mecanismos intracelulares muy similares.


Aunque hasta la fecha, sólo tres receptores opioides han sido clonados (DOP, KOP y MOP), al menos 13 subtipos diferentes de receptores opioides fueron identificados utilizando métodos farmacológicos por más de 10 años. Continúan las investigaciones para descubrir la razón de esta discrepancia. Explicaciones postuladas son:
- Las variantes de empalme del receptor (sin embargo, la expresión es baja en el sistema nervioso central y el dominio C-terminal intracelular del receptor se ve más afectado que la unión al ligando extracelular N-terminal).
- Receptores opioides funcionales heterodimericos como DOP-KOP y DOP-MOP, podrían existir.
- Los ligandos dirigidos por señales de proteína G, podrían producir diferentes efectos en sistemas de segundos mensajeros como la β-arrestina, es decir agonismo parcial.
EVENTOS INTRACELULARES
Una vez que el ligando se ha unido al receptor opioide, la proteína G asociada es activada. La subunidad α intercambia el GDP unido por GTP, y la subunidad βγ se disocia, y es libre para interactuar con el sistema de segundos mensajeros y los canales iónicos. Con la unión al receptor opioide clásico, hay una disminución en la producción de monofosfato de adenosina cíclico (cAMP), debido a que la adenilato ciclasa es inhibida, y también la conductancia al potasio esta aumentada, con una reducción en la conductancia de calcio a través de la membrana celular. Esto causa hiperpolarización celular, y excitabilidad neuronal reducida con liberación disminuída de neurotransmisores. Este es un mecanismo sostenible para los efectos clínicos de los opioides, pero es sorprendentemente no probado hasta la fecha.
Otros sistemas de segundos mensajeros están acoplados a la activación de los receptores opioides, como la proteína kinasa activada por mitógenos (MAP), y la fosfolipasa C que media la cascada que conduce a la formación de inositol trifosfato y diacil glicerol.
El concepto de señalización de proteína G acoplada a receptor ligando dirigido (PGAR) ha sido recientemente propuesto (figura 1). La activación de PGAR puede conducir a cualquiera de los dos, señalización igual/ imparcial o señalización desigual/parcial a través de proteína G o β- arrestina mediando las vías de señalización intracelular.
La implicancia es que la analgesia y los efectos adversos pueden ser traducidos diferencialmente por esas dos vías. En la β-arrestina de ratones “knockout” el efecto anti nociceptivo de la morfina fue mejorado y prolongado, mientras que la depresión respiratoria, la constipación y los efectos de abstinencia producidos por la Naloxona, fueron atenuados.
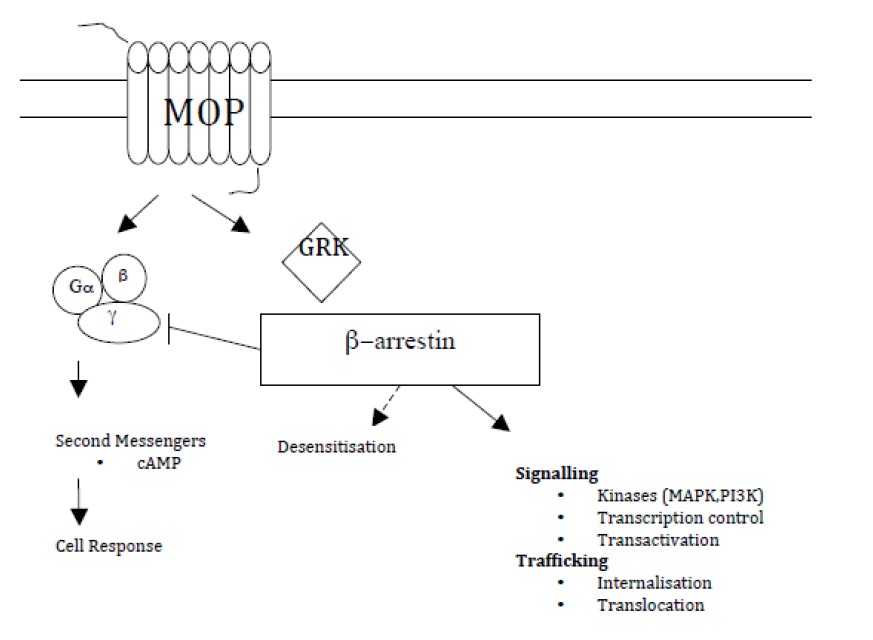
Figura 1. Receptor opioide y cascada intracelular. Proteina kinasa activada por mitogeno (MAPK), fosfoinositol 3- kinasa (PI3K), proteína G acoplada a receptor de kinasa (GRK)
ADMINISTRACION DE OPIOIDES A LARGO PLAZO
La exposición prolongada a opioides conduce a múltiples adaptaciones en los sistemas de señalización de los segundos mensajeros, lo cual podría ser responsable de la tolerancia, sensibilización, y síntomas de abstinencia.
Las proteínas kinasas intracelulares son las responsables de la fosforilación aguda de los receptores opioides MOP y los DOP, lo que resulta en tolerancia a los efectos de los agonistas.
La internalización de los receptores es común a todas las proteínas G asociadas a receptores, y está controlada por mecanismos diferentes a la interacción agonista receptor. Las proteínas G acopladas a receptores de kinasas, fosforilan a los receptores unidos a agonistas, promoviendo la interacción con las β- arrestinas, la cual interfiere con la proteína G acoplada, y promueve la internalización del receptor. La internalización del receptor puede tener respuestas divergentes como degradación del receptor, causando pérdida de la función o desfosforilación del receptor y reciclaje hacia la superficie celular, con mejora de la señalización.
La superactivación de la adenil ciclasa ocurre con la administración crónica de opioides agonistas. La alteración en los niveles de AMPc trae consigo numerosos cambios secundarios.
Hiperalgesia inducida por opioides
La HIO es una respuesta paradójica a los agonistas opioides por lo cual en lugar de que ocurra un efecto analgésico, o antinociceptivo, hay un aumento en la percepción del dolor.
Los mecanismos por los cuales los opioides inducen hiperalgesia incluyen:
- Regulación en alza de neurotransmisores exitatorios tales como la sustancia P y PGAR en fibras aferentes primarias en la medula espinal.
- Aumento de la liberación evocada de transmisores exitatorios en la médula espinal.
- Regulación en alza de los niveles de dinorfina, promoviendo que aumente la contribución de los nociceptores aferentes.
- Activación de la pan facilitación de la medula rostroventral.
- Aumento de la colecistoquinina (CCK) en el tallo cerebral que actúa a través de las vías descendentes.
- Los antagonistas del receptor TRPV1 han demostrado que revierten HIO.
- Los mecanismos de receptores de NMDA en relación con la sensibilización central.
- La activación glial a través de receptores toll like 4 (TLR4)
TERAPIA DUAL DE OPIOIDES
Las combinaciones de analgésicos a menudo producen efectos farmacológicos mayores que la suma de los dos. Esto se denomina sinergia. La metadona y morfina demostrado la sinergia en modelos animales de analgesia. Curiosamente los efectos sobre el tránsito gastrointestinal no mostraron sinergia.
GENETICA
Las variantes de empalme
Un solo gen (OPMR1) se ha asociado con MOP. Los genes consisten en exones e intrones. En situaciones normales, los intrones son empalmados para que los exones combinados puedan ser transcriptos en ARNm, y luego traducidos a proteínas receptoras. Splicing alternativo (variante de empalme) es una manera en la que un gen único puede producir una gran variedad de proteínas diferentes. Los ratones que carecen del exón 1 de MOP fueron insensibles a la morfina y aquellos que carecen del exón 2 eran sensibles a la morfina, pero no tenían una respuesta antinociceptiva de diamorfina (heroína), fentanil o morfina-6-glucurónido. La relevancia de estos hallazgos a la variabilidad clínica no está clara en la actualidad.
La genética entra en el juego cuando se considera el metabolismo de los opioides, y la codeína es un caso interesante. La codeína, puede ser considerada una pro-droga y esta metabolizada por tres vías hepáticas. El producto de la citocromo P3A4 (CYP3A4) es la norcodeína, mientras que la codeína-6-glucuronido es producida por UGT 2B7 (cerca del 80% del metabolismo de codeína). Se ha postulado a la codeína-6-glucuronido como la responsable del efecto analgésico de codeína, pero la vía CYP2A6 esta extensamente aceptada como la más importante (a pesar de ser responsable de menos del 5% del metabolismo de codeína). El producto de la vía CYP2A6 es la morfina. Hay varios fenotipos de CYP2A6 de metabolizadores pobres, donde la codeína carece de eficacia, a metabolizadores ultra-rápidos, donde esta incrementada la formación de morfina, conduciendo a altos riesgos de toxicidad. Esto, ha sido demostrado en el reporte de un caso de un infante con lactancia materna, el cual falleció debido a toxicidad por opioide. La madre era una metabolizadora ultra-rápida que tomaba 60mg de codeína por día.
DIMERIZACION DEL RECEPTOR
Los receptores opioides existen como entidades únicas, pero pueden existir como homodimeros tales como KOP-KOP, o MOP-MOP, y heterodimeros como DOP-MOP, y DOP-KOP. Esta dimerización altera las propiedades farmacológicas, y la afinidad altamente selectiva para agonistas y antagonistas se reduce. Los agonistas parcialmente selectivos y los opioides endógenos tienen elevada afinidad por esos complejos dimericos. Aquellos heterodiméricos podrían explicar la variabilidad en las propiedades moleculares y farmacológicas de los receptores opioides.
ALGUNOS OPIOIDES ESPECIFICOS
Morfina
La morfina es un derivado fenantrenico, por lo que es agonista de los receptores MOP y KOP. Junto a la codeína ha sido publicada en la lista WHO de medicinas esenciales en marzo de 2011. Puede que la codeína sea removida de la lista WHO en la próxima edición, y está sujeta a revisión. Su biodisponibilidad es del 15-50%, debido a un extenso metabolismo en el primer pasaje hepático. Se encuentra unido a proteínas en un 20-40%, predominantemente albumina, y su volumen de distribución (VD) es de 3.4-4.7 l/kg. El grado de analgesia y la concentración plasmática no está claramente relacionada. El metabolismo ocurre en el hígado a morfina-3-glucuronido, morfina-6-glucuronido, y normorfina. M-6-G produce efectos analgésicos, y M-3-G efectos exitatorios. La excreción ocurre predominantemente por orina como un conjugado glucuronido. 7-10% aparece en materia fecal como morfina conjugada. El aclaramiento es de 12-23ml/ml/kg, y la vida media de eliminación es de 1.7-4.5 hs. El efecto pico analgésico ocurre a los 30-60 minutos después de su administración parenteral, debido a su baja solubilidad en lípidos (transito lento a el sistema nervioso), y la duración de su efecto es de 3-4 horas.
Hidromorfona
La hidromorfina tiene farmacocinética y duración de acción similar a la morfina, pero es 5 veces más potente, con un comienzo de acción ligeramente más rápido. La glucuronización únicamente ocurre en la posición 3, siendo mejor tolerado en pacientes con fallo renal debido a la ausencia de un metabolito activo.
Fentanilo, remifentanilo y alfentanilo
Estos opioides son agonistas MOP, los cuales son muy usados en el periodo perioperatorio. Ellos muestran diferencias farmacocinéticas incluyendo elevada solubilidad en lípidos comparada con morfina, resultando en rápido comienzo de acción y rápida finalización del efecto. Remifentanilo tiene una corta vida media sensible al contexto, debido a su metabolismo por esterasas tisulares y plasmáticas no especificas. Remifentanilo ha sido usado para generar un modelo experimental de hiperalgesia.
Buprenorfina
La buprenorfina es un derivado sintético del alcaloide tebaína. Actúa como un agonista parcial de los receptores MOP, y se disocia lentamente produciendo una analgesia prolongada comparada con morfina. Tiene alta afinidad, pero baja actividad intrínseca a los receptores KOP. Debido a su significativo primer paso hepático, se prefiere la vía sublingual. Su biodisponibilidad es altamente variable, incluso por vía intramuscular, entre el 40-90%. Se encuentra unida a proteínas en un 96% in vitro, y su VD es de 3.2l/kg. Se metaboliza en el hígado por dealquilación con subsecuente conjugación o glucuronidos; los conjugados polares parecen ser excretados en la bilis e hidrolizados por bacterias en el tracto gastrointestinal. Es excretada preferentemente por materia fecal como buprenorfina y derivados dealquilados. El aclaramiento es de 1l/min. La vida media de eliminación es de 5 horas.
Como es un agonista parcial, la buprenorfina puede antagonizar los efectos de la morfina, y puede precipitar un síndrome de abstinencia en pacientes con dependencia de opioides.
Metadona
La metadona es un opioide sintetico desarrollado en 1942. Es una droga básica y lipofilica (pKa 9.2) y existe como una mezcla racémica de dos enantiomeros, R-metadona y S- metadona. La R-metadona es un agonista potente de los receptores MOP y DOP. El enantiomero de la S-metadona es inactivo como agonista para el receptor MOP, pero actua como antagonista de los receptores NMDA. En la administración por vía oral, el tiempo para alcanzar el pico plasmático es de 2.5-3 horas. Su biodisponibilidad por vía oral es alta, cerca del 85%. El VD es elevado en humanos 4.2-9.2l/kg. A pH fisiológico el 86% de la metadona está unida a proteínas plasmáticas, principalmente la alfa-1- glicoproteína acída. A diferencia de la morfina, la metadona es biotransformada en el hígado antes de ser conjugada, y con una dosis diaria menor a 55mg la mayoría de los metabolitos son aclarados en las heces. Metadona es metabolizada por la enzima citocromo P-450. La enzima principal responsable de la N- detilación de la metadona es CYP3A4, con menor participación la CYP1A2 y la CYP2D6. El producto principal del metabolismo es el 2-etileno-1,5-dimetil-1,3,3- difenilpirrolidina (EDDP), y es inactivo. Hay grandes variaciones interindividuales en la farmacología de la metadona. La excreción renal es variable y pH dependiente, con excreción aumentada cuando el pH urinario desciende. La eliminación de la metadona es bifásica. La fase α es de 8-12 horas, y la fase β de eliminación es aún más prolongada de 30-60 horas. A pesar de su prolongado periodo de eliminación, la duración de la analgesia es 8-12 horas, y para el manejo del dolor la dosis diaria es dividida en 2 o 3 administraciones. Existe un riesgo real de acumulación y toxicidad con dosis repetidas.
Tramadol y Tapentadol
Tramadol es un agonista parcial MOP con una acción adicional, inhibe la recaptación de serotonina y noradrenalina, y Tapentadol es un agonista MOP con inhibición de la recaptación de noradrenalina. Tramadol es metabolizado a un metabolito activo M1, el cual tiene mayor afinidad por MOP que el compuesto original.
RESUMEN
No hay un único mecanismo adecuado que explique la variabilidad intraindividual o interindividual observada con los opioides. La evidencia disponible sugiere que una constelación de factores neurobiológicos, demográficos, médicos y del paciente, contribuyen todos a determinar la respuesta del paciente a opioides particulares. Los opiodes continúan siendo el componente clave del manejo del dolor agudo, y en dolor oncológico. El uso de opioides en dolor crónico de causa no maligna es limitado por la tolerancia y la hiperalgesia.

RESPUESTAS A LAS PREGUNTAS
- a.MOP
- c.30-60 minutos
- a. verdadero b.verdadero c.verdadero d.falso e.verdadero
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/