Paediatric Anaesthesia

PUNTOS CLAVE
- Las enfermedades mitocondriales son diversas, pueden afectar un amplio rango de órganos y presentarse con una mulRtud de síntomas.
- El manejo perioperatorio involucra minimizar el stress metabólico de la cirugía y el riesgo de una “encefalopaWa metabólica”, a la que a menudo contribuyen el ayuno o una enfermedad intercurrente.
- Las metas intraoperatorias incluyen mantenimiento de la temperatura central, glucosa sanguínea, perfusión de órganos y oxigenación.
- Los agentes inhalatorios son seguros para usar en pacientes con enfermedades mitocondriales, aunque algunos pacientes pueden tener una sensibilidad aumentada.
- Las infusiones de Propofol deberían probablemente ser evitadas en pacientes con enfermedades mitocondriales, sin embargo, dosis únicas en bolo para inducción de la anestesia generalmente son bien
INTRODUCCIÓN
Las enfermedades mitocondriales comprenden un diverso grupo de desórdenes que afectan adversamente la función de la mitocondria. La primera enfermedad mitocondria fue identificada hace 30 años, y ahora hay casi 300 mutaciones genéticas conocidas.1 Este artículo revisará la biología básica de la mitocondria, las enfermedades mitocondriales y sus implicaciones anestésicas.
Biología Mitocondrial
Las mitocondrias son organelos de células eucarióticas que pueden haber sido antiguas formas de vida independientes que simbióticamente se fusionaron con bacterias hace casi 2 millones de años.1 Poseen su propio DNA y están ubicuamente presentes en todas las células excepto los eritrocitos. Tienen múltiples membranas y dobleces que llevan a cabo funciones especializadas. Las proteínas mitocondriales están codificadas tanto por DNA nuclear (herencia Mendeliana) como por DNA mitocondrial (herencia materna); por lo que, las enfermedades mitocondriales pueden ocurrir como resultado de mutaciones del DNA nuclear y/o mitocondrial.2 Las mitocondrias son conocidas como la “central de energía ATP de la cèlula”. Su función primaria es la producción de trifosfato de adenosina (ATP) via fosforilación oxidativa, por la que electrones son pasados entre los diferentes complejos de la cadena de transporte de electrones (Figura 1) 2,4 Las
Mitocondrias también son importantes en otras vías metabólicas incluyendo el Ciclo de Kreb’s, el ciclo de la urea, y la oxidación de ácidos grasos-beta.3,4
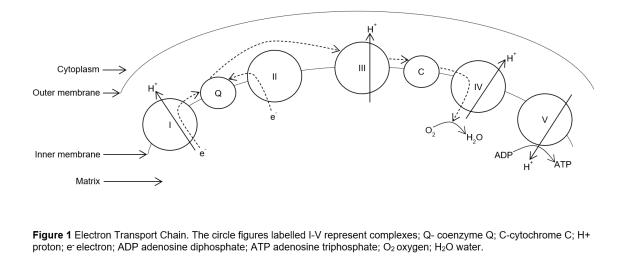
Figura 1 Cadena de Transporte de electrones. Las figuras circulares rotuladas I-V representan complejos; Qcoenzima Q; C- citocromo C; H+ protón; e- electrón; ADP difosfato de adenosina; ATP trifosfato de adenosina; O2
Oxígeno; H2O agua.
Cytoplasm: citoplasma; Outer Membrane: membrana externa; Inner Membrane: membrana interna; Matrix:
matriz.
ENFERMEDAD MITOCONDRIAL
Las enfermedades mitocondriales tienen una prevalencia estimada de 1 en 4000, y en la mayoría de casos se manifiestan alrededor de la edad de 20 años.5 Enfermedades mitocondriales resultantes de mutaciones de DNA mitocondrial demuestran heteroplasmia y niveles umbral. Heteroplasmia es el fenómeno por el cual cada célula puede albergar una mezcla de mitocondrias normales y mutantes, y nivel umbral es el fenómeno por el cual una cierta cantidad de mitocondria mutante necesita estar presente antes que la célula empiece a mostrar disfunción (Figura 2). Esto significa que mutaciones idénticas entre miembros de una familia pueden tener genotipos clínicos variables.2,6
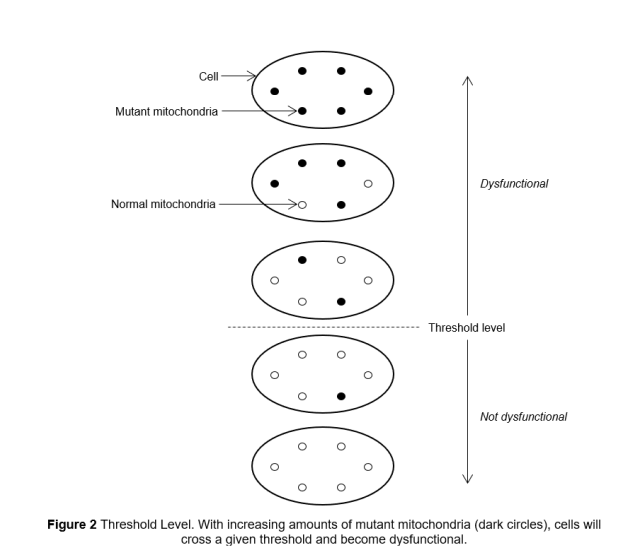
Figura 2 Nivel Umbral. Con cantidades crecientes de mitocondrias mutantes (círculos oscuros), las células van a atravesar un umbral dado y se volverán disfuncionales. Cell: célula; Mutant Mitochondria: Mitocondria Mutante; Normal Mitochondria: Mitocondria Normal; Dysfunctional: Disfuncional; Threshold level: Umbral; Not dysfunctional: No disfuncionales.
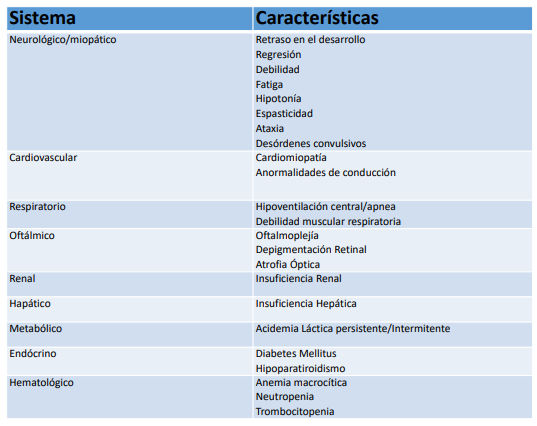
Tabla. Características Clínicas de la Enfermedad Mitocondrial. De Wallace et al2 con permiso de Wiley por parte
de Paediatric Anaesthesia. Esta imagen/contenido no está cubierta por los términos de la licencia de Creative
Commons de esta publicación. Para permiso de reutilizar, favor contactar al dueño de los derechos de autor.
Frecuentemente se refiere a las enfermedades mitocondriales como desórdenes que pueden ‘afectar cualquier sistema, con cualquier síntoma, y por cualquier modo de herencia’.1 Los órganos y tejidos que tienen una alta rotación de ATP están afectados proporcionalmente. En general, un paciente con una miopatía o encefalopatía con un nivel de lactato elevado debería traer a consideración la posibilidad de un defecto mitocondrial.6 Adicionalmente, múltiples sistemas de órganos pueden estar afectados (Tabla).2 La clasificación de enfermedades mitocondriales es variada basada ya sea en anormalidades
específicas de complejos de cadena de transporte de electrones, mutaciones de DNA mitocondrial y/o nuclear o en fenotipos clínicos. La clasificación clínica incluye síndromes tales como MELAS (encefalopatía mitocondrial, acidosis láctica y episodios de pseudo-infartos cerebrales), MINGIE (encefalopatía neurogastrointestinal mitocondrial, MERRF (epilepsia mioclónica con fibras rojas desgarradas), Síndrome de Kearns-Sayre y Síndrome de Leigh. Hay, sin embargo, un gran traslape y ninguna correlación clara entre los hallazgos clínicos y el sitio del defecto bioquímico.7 La mejor prueba
diagnóstica es una biopsia de músculo; sin embargo, esto se lleva a cabo solo si la prueba genética es equívoca. Las biopsias de músculo son de preferencia hechas en el Vasto Lateral, y los hallazgos característicos de fibras rojas desgarradas o fibras citocromo c-oxidasa negativo son a menudo buscados en la tinción histoquímica. Otras características bioquímicas de apoyo incluyen niveles elevados de lactado en sangre, orina o líquido cerebro espinal. Características de apoyo en Neuroimágenes incluyen lesiones de pseudo-infarto cerebral en distribuciones no-vasculares; enfermedad difusa de materia blanca; involucración bilateral de núcleos de la materia gris profunda en los ganglios basales, mesencéfalo y/o tallo cerebral; y el doble pico del lactato en la espectroscopía de resonancia magnética cerebral.8
No hay una cura conocida para las enfermedades mitocondriales, y el tratamiento es en gran medida de soporte. Esto incluye enfocarse en la optimización de la producción de energía, reducción de las pérdidas de energía, evitar las toxinas, aliviar los síntomas y monitoreo de las complicaciones. Se ha demostrado que el ejercicio mejora los síntomas y la fuerza, y que aumenta el contenido mitocondrial y la captación de oxígeno. Los suplementos nutricionales se prescriben comúnmente para reemplazar las deficiencias que puedan ocurrir cuando la función mitocondrial falla. Consiste de múltiples vitaminas y
cofactores incluyendo coenzima Q10, ácido alfa-lipoico, L-carnitina, creatina y ciertas vitaminas del grupo B. La evidencia que apoya el uso de la mayoría de estos suplementos es limitada.8
IMPLICACIONES ANESTESICAS
Preoperatorias
Los pacientes con enfermedades mitocondriales a menudo tienen procedimientos diagnósticos y
terapéuticos en quirófano, tales como biopsias musculares, escaneos de imagenología por resonancia
magnética/tomografía computarizada, endoscopías, elaboración de gastrostomías y cirugía de
estrabismo. Durante la evaluación preoperatoria, el anestesiólogo debe comprobar el grado de
compromiso neurológico y muscular con evidencia de involucramiento cardiorrespiratorio. Las
características a buscar en la historia y examen físico son la severidad de la fatigabilidad, disfagia,
hipoventilación, apnea, cardiomiopatía, y/o bloqueo cardíaco. Las investigaciones preoperatorias en
gran medida dependen de la severidad de la enfermedad, los sistemas de órganos afectados y la cirugía
requerida. Exámenes de sangre basales útiles incluyen una cuenta hemática completa, electrolitos,
creatinina, urea, pruebas de función hepática, glucosa sanguínea, lactato, piruvato y creatin cinasa (CK
o CPK). Otras investigaciones útiles pueden incluir espirometría, R-x de tórax, gases sanguíneos,
electrocardiograma y ecocardiograma. Es de suma importancia que estos pacientes no ayunen por un
período de tiempo prolongado.2,6,8 En nuestra institución, usamos maltodextrina ya sea mezclada con
agua o jugo de manzana sin pulpa hasta 2 horas previo a la inducción para prevenir la hipoglicemia. La
maltodextrina es un polisacárido de fácil digestión que puede ser considerado un líquido claro. Por
último, consultas multidisciplinarias pueden ser requeridas ya que los pacientes a menudo son tratados
por una variedad de especialistas médicos.7
Intraoperatorios
El objetivo principal en pacientes con enfermedades mitocondriales es minimizar el stress metabólico
de la cirugía y el riesgo de una “encefalopatía metabólica”, que a menudo es desencadenada por una
enfermedad intercurrente (o concomitante) y/o ayuno. Es importante evitar la hipotensión
intraoperatoria, la hipoxia, hipoglicemia e hipotermia.6,9 Para mantener la homeostasis de la glucosa,
soluciones conteniendo glucosa deben ser administradas con mediciones regulares de la glucosa
sanguínea. La excepción a esta regla es en pacientes con desórdenes del metabolismo del piruvato o
dietas cetogénicas para control de las convulsiones; tales pacientes deben recibir glucosa administrada
con precaución y ser monitoreados para asegurarse que no desarrollan hiperglicemia y/o acidosis
láctica.6,8 Las soluciones conteniendo lactato tales como los compuestos lactato de sodio/solución de
Ringer es mejor evitarlas, y las mediciones intraoperatorias del lactato pueden ser un marcador útil del
stress metabólico.10,11
Postoperatorios
Una buena analgesia postoperatoria es importante para estos pacientes ya que la respuesta al dolor de
la cirugía puede empeorar la acidosis láctica. Como resultado, un abordaje analgésico multimodal debe
ser usado. Las técnicas regionales deben usarse si ello es posible y práctico.2
PERFIL DE SEGURIDAD FARMACOLÓGICO
Han surgido preocupaciones respecto a la seguridad de diferentes fármacos utilizados en anestesia.
Casi todos los fármacos demuestran evidencia bioquímica de inhibición mitocondrial directa, pero las
implicaciones clínicas de ello son menos claras. La siguiente sección revisará brevemente los perfiles de
seguridad de cada una de las principales clases de fármacos anestésicos.
Anestésicos Volátiles
Se ha demostrado en estudios in vitro que los agentes anestésicos volátiles inhiben el complejo I de la
cadena de transporte de electrones (Figura 1). Los estudios correlacionando este efecto bioquímico a
uno clínico han sido escasos. Un estudio observando a 16 niños con enfermedad mitocondrial
demostrada por biopsia encontró que 1 paciente con una mutación del complejo I y otro con
enfermedad de Leigh pueden tener una sensibilidad aumentada a la anestesia volátil.3 A pesar que no
han habido estudios posteriores para elucidar esta interrogante, una revisión retrospectiva ha
demostrado el uso de rangos clínicos “normales” de concentraciones inspiratorias de sevofluorane sin
daños.4
La preocupación acerca de la asociación de enfermedades mitocondriales e hipertermia maligna
probablemente empezó en 1985. Un reporte único de un caso en Japón describía a un niño de 2 años
con enfermedad mitocondrial que desarrolló rigidez muscular, hiperkalemia e hipertermia posterior a
una anestesia general que incluyó halothane y suxametonio.12 A pesar del amplio uso de agentes
volátiles, no han habido más reportes de casos de hipertermia maligna en pacientes con enfermedades
mitocondriales. Más aún, la Asociación de Hipertermia Maligna de los Estados Unidos (MHAUS)
recomienda que los agentes volátiles no deberían ser evitados solo por la preocupación de una posible
susceptibilidad a hipertermia maligna.13
Anestesia Intravenosa
El Propofol se ha demostrado que inhibe múltiples complejos de la cadena de transporte de electrones y el transporte de ácidos grasos libres (FFA) a través de las membranas mitocondriales.14 El Propofol fue probablemente usado más comúnmente como parte de una anestesia no desencadenante a principios de los 1990’s, cuando se pensaba que estos pacientes eran susceptibles a hipertermia maligna. Sin embargo, la preocupación empezó a surgir aproximadamente hace 20 años, con reportes de casos que sugerían que los pacientes con síndrome de infusión de propofol (PRIS)15 tenían anormalidades bioquímicas similares a las de las enfermedades mitocondriales.16 Reportes de casos posteriores han
descrito el desarrollo de PRIS en un paciente con una deficiencia adquirida de carnitina17 y otros con enfermedad mitocondrial18,19 a quienes se les administró infusiones de Propofol. La patofisiología del PRIS permanece poco clara; sin embargo, la evidencia sugiere defectos mitocondriales en la producción de ATP como la causa más probable.20 Algunos autores han sugerido que los pacientes que desarrollan PRIS pueden tener formas subclínicas de enfermedades mitocondriales,11 y otros han recomendado que los pacientes que desarrollan PRIS sean investigados para estas enfermedades.18 La evidencia acerca del propofol es conflictiva: una revisión reciente sugiere que a pesar que los bolos de propofol son probablemente seguros en las enfermedades mitocondriales, las infusiones podrían no serlo.14 Nuestra opinión, de la evaluación de la literatura publicada, es que el propofol no es el agente anestésico de elección; una cuidadosa valoración de los bolos de propofol en los pacientes sin formas severas de enfermedades mitocondriales y sin enfermedad crítica es probablemente seguro. SIn embargo las infusiones de propofol probablemente deban ser evitadas. El uso de Ketamina, dexmedetomidina y benzodiazepinas en pacientes con enfermedades mitocondriales no se ha asociado
en la literatura con daño.14
Bloqueadores Neuromusculares
Las preocupaciones rodeando los bloqueadores neuromusculares se relacionan principalmente a su
perfil farmacodinámico. Los bloqueadores neuromusculares despolarizantes como el suxametonio
deberían evitarse dado el riesgo de una respuesta hiperkalémica exagerada.14 Hay reportes conflictivos
acerca de si los pacientes con enfermedades mitocondriales tienen una sensibilidad aumentada a los
fármacos bloqueadores neuromusculares no-depolarizantes; a pesar que algunos estudios reportan
una sensibilidad aumentada comparado con la población general, otros no han mostrado diferencia.2
Se ha recomendado que “cualquier niño con hipotonía debe ser considerado a riesgo de una respuesta
variable a la relajación muscular y las dosis ajustadas de manera correspondiente”.21 Casi todas las
recomendaciones incluyen el uso de un neuroestimulador.2,5,10 La reversión del bloqueo
neuromuscular con neostigmina5 o sugamadex22-24 no se ha asociado con daño.
Anestésicos Locales
Estudios in vitro en tejido animal han demostrado que la bupivacaína inhibe el transporte de FFA
similar a propofol25; sin embargo, la evidencia de daño en humanos es escasa. Hay un único reporte de
caso de una bradiarritmia intraoperatoria luego de la infiltración subcutánea de ~0.3 mg/kg de
bupivacaína en un paciente con deficiencia de carnitina.26 Posterior a esto, no han habido más eventos
adversos reportados en humanos hasta donde llega el conocimiento del autor. Más aún, múltiples
instituciones reportan usar, sin daño, bupivacaína para biopsias musculares en pacientes con
enfermedades mitocondriales.4,27 Un artículo de revisión reciente sugiere que hay ventajas con el uso
de anestesia local, ya que provee analgesia sin los efectos represores respiratorios de los opioides; sin
embargo, no recomienda ningún agente sobre otro.14
Opioides
Los opioides no han sido implicados en tener efectos bioquímicos significativos sobre la mitocondria.14
No hay reportes de caso de daño asociado con su uso; sin embargo, generalmente se recomienda
precaución con respecto a sus efectos depresores respiratorios.2 El remifentanil es particularmente
ventajoso en este respecto, dado su perfil farmacocinético favorable.14
RESUMEN
La enfermedad mitocondrial es una enfermedad multisistémica compleja y relativamente común. Puede presentarse con una variedad de síntomas, y establecer un diagnóstico continúa siendo un desafío y potencialmente invasivo. El anestesiólogo puede verse involucrado en el cuidado de estos pacientes con procedimientos que son diagnósticos y/o terapéuticos. Es
importante identificar preoperatoriamente los efectos a órganos blanco de la enfermedad, minimizar el stress de la cirugía y estar consciente de los cuestionamientos potenciales con cada clase de fármaco anestésico.
RECONOCIMIENTO
Agradecemos a la Dra. Shanti Balasubramaniam (genetista metabólico, The Children’s Hospital en
Westmead) por su ayuda.
REFERENCIAS
1. Falk MJ, Medscape. Mitochondrial diseases: current state of understanding. https://www.medscape.com/
viewarticle/896188. Accessed December 2, 2019.
2. Wallace JJ, Perndt H, Skinner M. Anaesthesia and mitochondrial disease. Paediatr Anaesth. 1998;8:249-254.
3. Morgan PG, Hoppel CL, Sedensky MM. Mitochondrial defects and anesthetic sensitivity. Anesthesiology.
2002;96:1268-1270.
4. Driessen J, Willems, S, Dercksen, S, et al. Anesthesia-related morbidity and mortality after surgery for muscle
biopsy inchildren with mitochondrial defects. Paediatr Anaesth. 2007;17:16-21.
5. Wisley NA, Cook PR. General anaesthesia in a man with mitochondrial myopathy undergoing eye surgery.
Eur J Anaesthesiol. 2001;18:333-335.
6. Niezgoda J, Morgan PG. Anesthetic considerations in patients with mitochondrial defects. Paediatr Anaesth.
2013;23:785-793.
7. Shipton EA, Prosser DO. Mitochondrial myopathies and anaesthesia. Eur J Anaesthesiol. 2004;21:173-178.
8. Parikh S, Goldstein A, Koenig MK, et al. Diagnosis and management of mitochondrial disease: a consensus
statement from the Mitochondrial Medicine Society. Genet Med. 2015;17:689-701.
9. Farag E, Arglaious M, Narouze S, et al. The anesthetic management of ventricular septal defect (VSD) repair
in a child with mitochondrial cytopathy. Can J Anaesth. 2002;49:958-962.
10. Hoppe K, Lehmann-Horn F, Jurkat-Rott K, et al. Mitochondrial disorders. Anasth Intensivmed ¨ .
2017;58:S125-S133.
11. Farag E, DeBoer G, Cohen BH, et al. Metabolic acidosis due to propofol infusion. Anesthesiology.
2005;102:697-698.
12. Ohtani Y, Miike T, Ishitu T, et al. A case of malignant hyperthermia with mitochondrial dysfunction. Brain
Dev. 1985;7:249.
13. Malignant Hyperthermia Association of the United States. Does mitochondrial myopathy (MM) increase an individual’s susceptibility to malignant hyperthermia (MH)? https://www.mhaus.org/healthcare-professionals/
mhaus-recommendations/does-mitochondrial-myopathy-mm-increase-an-individuals-susceptibility-tomalignant-hyperthermia-mh/. Accessed December 1, 2019.
14. Hsieh VC, Krane EJ, Morgan PG. Mitochondrial disease and anesthesia. J Inborn Errors Metab Screen.
2017;5:1-5.
15. Monojit, P. Propofol Infusion Syndrome. Anaesthesia Tutorial of the Week. 2020:435. https://
resources.resources.wfsahq.org/atotw/propofol-infusion-syndrome-atotw-435/ (accessed on 3rd Nov 2020)
16. Wolf A, Weir P, Segar P, et al. Impaired fatty acid oxidation in propofol infusion syndrome. Lancet.
2001;357:606-607.
17. Uezono S, Hotta Y, Takakuwa Y, et al. Acquired carnitine deficiency: a clinical model for propofol infusion
syndrome? Anesthesiology. 2005;103:909.
18. Savard M, Dupre N, Turgeon AF, Desbiens R, Langevin S, Brunet D. Propofol-related infusion syndrome
heralding a mitochondrial disease: case report. Neurology. 2013;81:770-771.
19. Vanlander AV, Jorens PG, Smet J, et al. Inborn oxidative phosphorylation defect as risk factor for propofol
infusion syndrome. Acta Anaesthesiol Scand. 2012;56:520-525.
20. Hemphill S, McMenamin L, Bellamy MC, et al. Propofol infusion syndrome: a structured literature review
and analysis of published case reports. Br J Anaesth. 2019;122:448-459.
21. Ross AK. Muscular dystrophy versus mitochondrial myopathy: the dilemma of the undiagnosed hypotonic
child. Paediatr Anaesth. 2007;17:1-6.
22. Iwata K, Tanabe K, Sugiyama Y, et al. Anesthetic management for a patient with very-long-chain acylcoenzyme A dehydrogenase deficiency. J Anesth. 2012;26:957-958.
23. Syed F, Turner H, AlGhamdi F, et al. Anesthetic management of a patient with carnitine-acylcarnitine
translocase deficiency. J Med Cases. 2018;9:127-130.
24. Kynes JM, Blakely M, Furman K, et al. Multidisciplinary perioperative care for children with neuromuscular
disorders. Children. 2018;5:126.
25. Weinberg GL, Palmer JW, VadeBoncouer TR, et al. Bupivacaine inhibits acylcarnitine exchange in cardiac
mitochondria. Anesthesiology. 2000;92:523-528.
26. Weinberg GL, Laurito CE, Geldner P, et al. Malignant ventricular dysrhythmias in a patient with isovaleric
acidemia receiving general and local anesthesia for suction lipectomy. J Clin Anesth. 1997;9:668-670.
27. Schnabel RM, Marcus MA, Theunissen HM, et al. Anesthetic management for a child with mitochondrial
complex II deficiency. Paediatr Anaesth. 2008;18:802-803.
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/