General Topics

KEY POINTS
- The QT interval on the electrocardiogram reflects the time taken for the ventricle to depolarise and repolarise.
- Long QT syndrome is a conduction disorder characterised by prolongation and dispersion of ventricular repolarisation, making torsade de pointes (‘twisting of the points’) more likely.
- Both congenital and acquired forms are recognised.
- Symptoms include palpitations, syncope and sudden cardiac arrest. Asymptomatic cases are common.
- Drug-induced QT prolongation is common in the hospital setting, particularly in the perioperative period.
- Appropriate avoidance of drugs that can prolong the QT interval, together with careful selection of anaesthetic drugs and avoidance of arrhythmogenic triggers, helps to ensure an uneventful perioperative course.
- Immediate recognition and treatment of torsade de pointes may be life-saving. Treatments include intravenous magnesium, overdrive pacing, electrical cardioversion and defibrillation depending on haemodynamic stability.
INTRODUCTION
The prolongation of the ventricular repolarisation time and QT interval dispersion (measured as interlead variability of QT) are known risk factors for the development of malignant arrhythmic episodes and cardiac arrest.
The true prevalence of long QT syndrome (LQTS) remains unknown, as it is often undiagnosed. The prevalence of congenital LQTS is estimated to be close to 1:2000 live births,1 but if genotype-positive/phenotype-negative individuals are included, the
numbers would probably be considerably higher. Likewise, acquired LQTS without torsades de pointes (TdP) is more common than TdP events, which may further explain underreporting. In one retrospective single-centre study, 0.7% of all hospitalised patients had a QTc ≥500 ms, but less than 6% of these patients experienced a life-threatening arrhythmia.2
LQTS is significant in surgical patients because many perioperative drugs are known to lengthen the QT interval. It has been shown that at the end of surgery, 80% of patients experience a significant QTc prolongation, ΔQTc 23 ms (95% confidence interval [CI] 20-25 ms).3
DEFINITION
The QT interval reflects both the ventricular depolarisation and repolarisation times in the electrocardiogram (ECG) and is measured from the onset of the QRS complex until the end of the T wave (Figure 1). Since the QT interval varies inversely with the heart rate (longer at slower rates and shorter at faster rates), the QT duration must be corrected for the heart rate (QTc). The simplest and most commonly used formula was developed by Bazett: QTc (ms) = QT interval (ms): = √ interval (ms).4
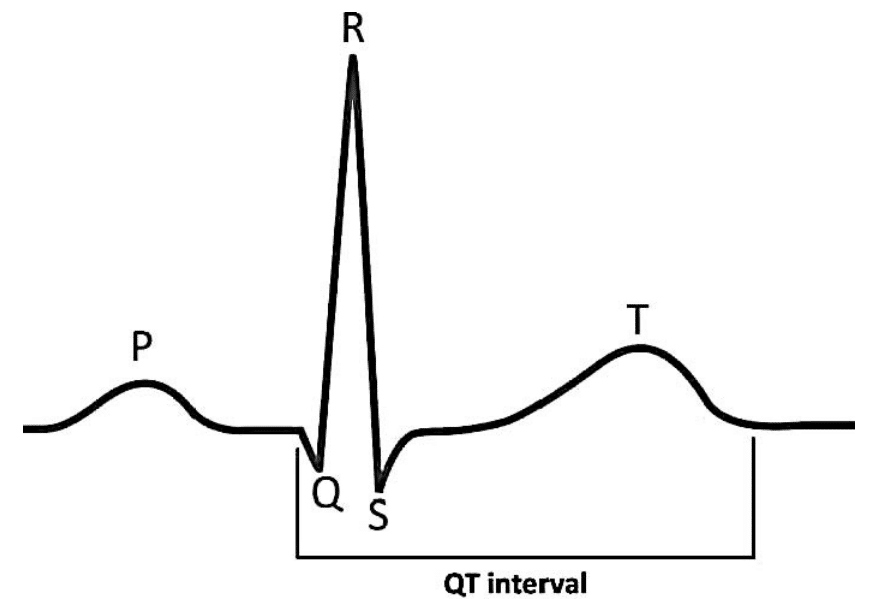
Figure 1. QT interval.
The QTc in healthy subjects varies widely, with a mean and standard deviation of 412 ± 25 ms, respectively, and a 99th percentile value of 470 ms in the adult population.5 A QTc interval greater than 480 ms is indicative of an LQTS.6 However, several limitations must be considered:
- The Bazett formula is less accurate at heart rate extremes, resulting in undercorrection at low heart rates and overcorrection at high heart rates.7
- Accurate measurements of the QT interval can be technically difficult, especially regarding the termination of the T wave and in cases of irregular rhythms. Therefore, the QT interval must be measured manually in several leads (eg, II and V5), averaged over different beats and adjusted for the heart rate.
- U waves should not be included, as they lead to overestimation of QTc.8
- It has been suggested that using a threshold of 500 ms for a prolonged QTc or using the JT interval (JT = QT-QRS) rather than the QT interval are more appropriate approaches when QRS prolongation is present.7
PATHOPHYSIOLOGY
An understanding of the normal myocardial action potential (Figure 2) is required to understand the pathophysiology behind QT prolongation:
- Phase 4, or the resting membrane potential, is when the cell membranes during diastole are polarised at –90 mV. This is mainly due to the constant outward leak of potassium ions through inward rectifier potassium (or IK1) channels.
- Phase 0 is the phase of rapid depolarisation. The membrane potential reaches þ20 mV due to a rapid inward flow of sodium ions through inward sodium (or INa) channels.
- Phase 1 is due to initial repolarisation caused by potassium ion efflux via transient outward potassium (or Ito) channels.
- Phase 2, or the plateau phase, represents an equilibrium between the entry of calcium ions into the cell via long-lasting and transient calcium (or ICa-L and ICa-T) channels, and the outward potassium currents via rapidly and slowly delayed rectifier (or IKr and IKs) channels.
- Phase 3 is the phase of rapid repolarisation where outward potassium currents (IKs and IKr) dominate.
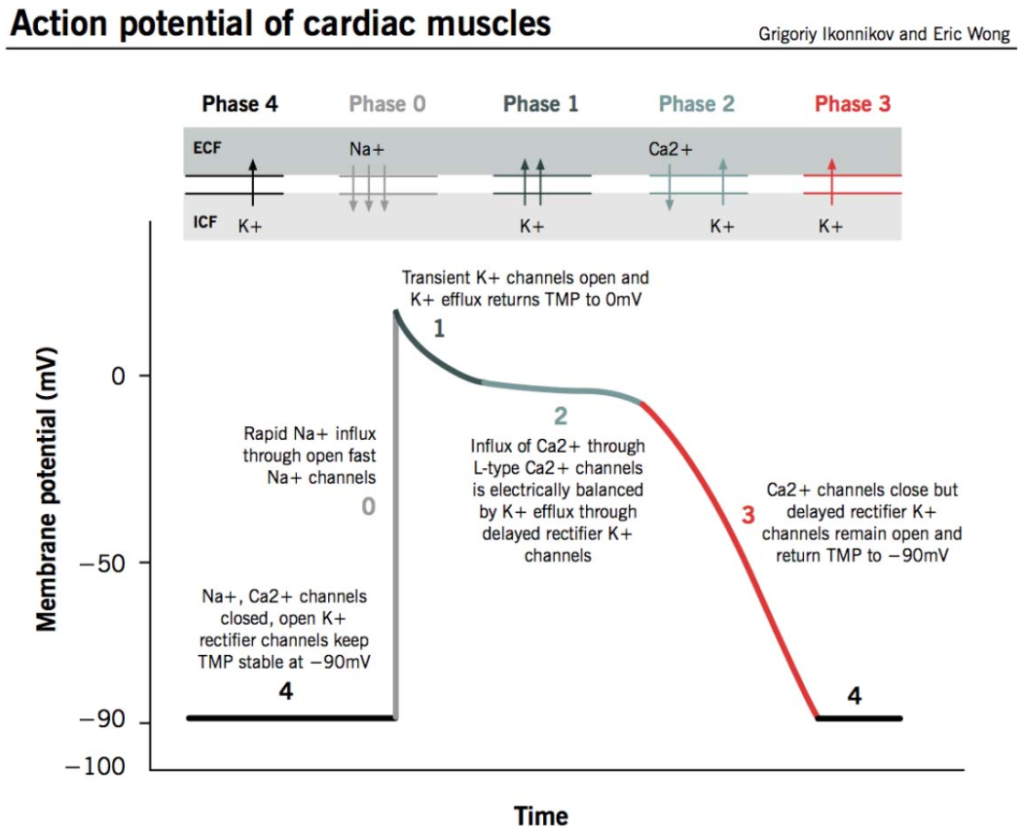
Figure 2. Myocardial action potential. TMP indicates transmembrane potential.13 Permission granted by Eric K. C. Wong, MD. This image/content is not covered by the terms of the Creative Commons license of this publication. For permission to reuse, please contact the rights holder.
Prolongation of the QT interval, as seen in LQTS, results from prolongation of repolarisation mainly in phase 3. This increases the propensity for early after-depolarisations, some of which reach threshold potential and can lead to TdP.
AETIOLOGY
The prolongation of repolarisation can be due to congenital mutations or acquired factors.
Congenital LQTS
Mutations in more than 15 genes have been identified in patients with congenital LQTS. Mutations in the KCNQ1, KCNH2 and SCN5A genes account for 75% to 80% of all congenital LQTS. Mutations in these genes lead to suppression of IKs channels, suppression of IKr channels and increased number of INa channels, respectively. Figure 3 illustrates several gene mutations in the context of the specific current perturbed.
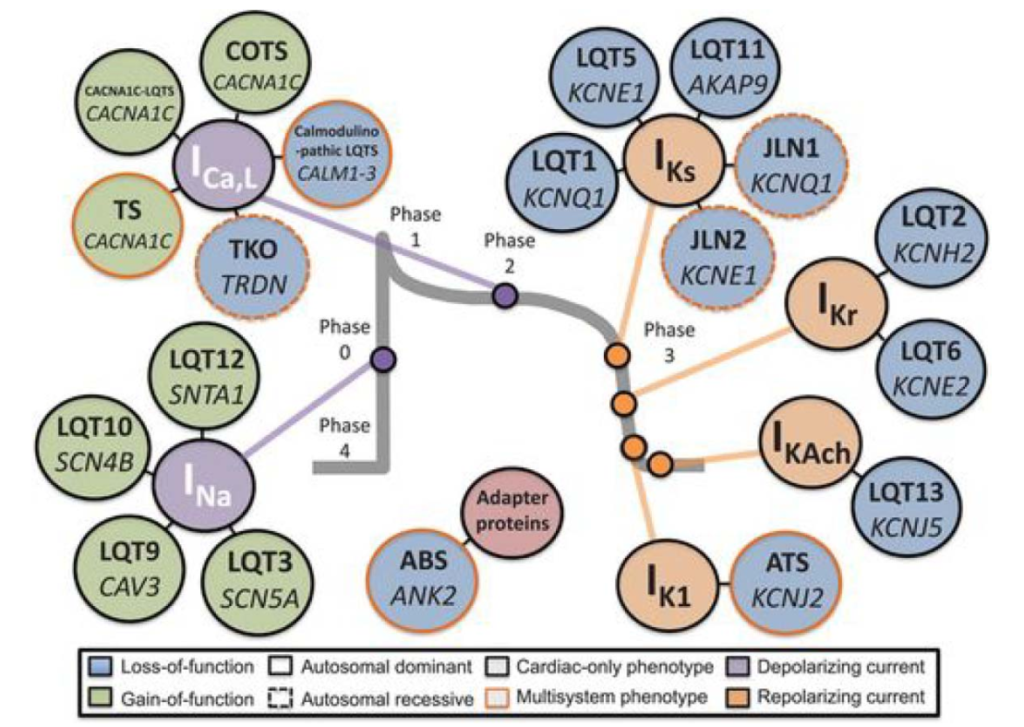
Figure 3. Current-centric classification of long QT syndrome genotypes.14 Reprinted from Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Translational Research, 161(1):1–14. Copyright 2013, with permission from Elsevier.
In general, 2 syndromes characterised by congenital LQTS have been described9:
- The more common autosomal dominant form, Romano-Ward syndrome, has only cardiac manifestations.
- The autosomal recessive form, Jervell and Lange-Nielsen syndrome, is characterised by both LQTS and sensorineural deafness.
Patients with multiple mutations exhibit significantly longer QT intervals and have a greater risk for life-threatening cardiac arrhythmias.
Acquired LQTS
Acquired LQTS usually requires exposure to drugs or electrolyte disturbances that induce an iatrogenic prolongation of the myocardial repolarisation time. Most drugs that lead to LQTS block the IKr current.10 In these patients, ventricular tachycardia usually follows short-long RR intervals (a premature ventricular contraction followed by a compensatory pause) or is associated with bradycardia. In fact, lower heart rates result in less potassium moving out of the cell during repolarisation, and the reduction in extracellular potassium further contributes to the drug-induced inhibition of IKr.10
Overlap may be present, in which some patients with acquired LQTS have a forme fruste of congenital LQTS, a mutation or polymorphism that is clinically unapparent until exposure to an additional insult occurs. QT prolongation often goes unnoticed until symptoms occur, so that acquired LQTS without TdP is more frequent than TdP events. Table 1 includes common causes and triggers of LQTS. A more complete list of such drugs is available at http://crediblemeds.org.11
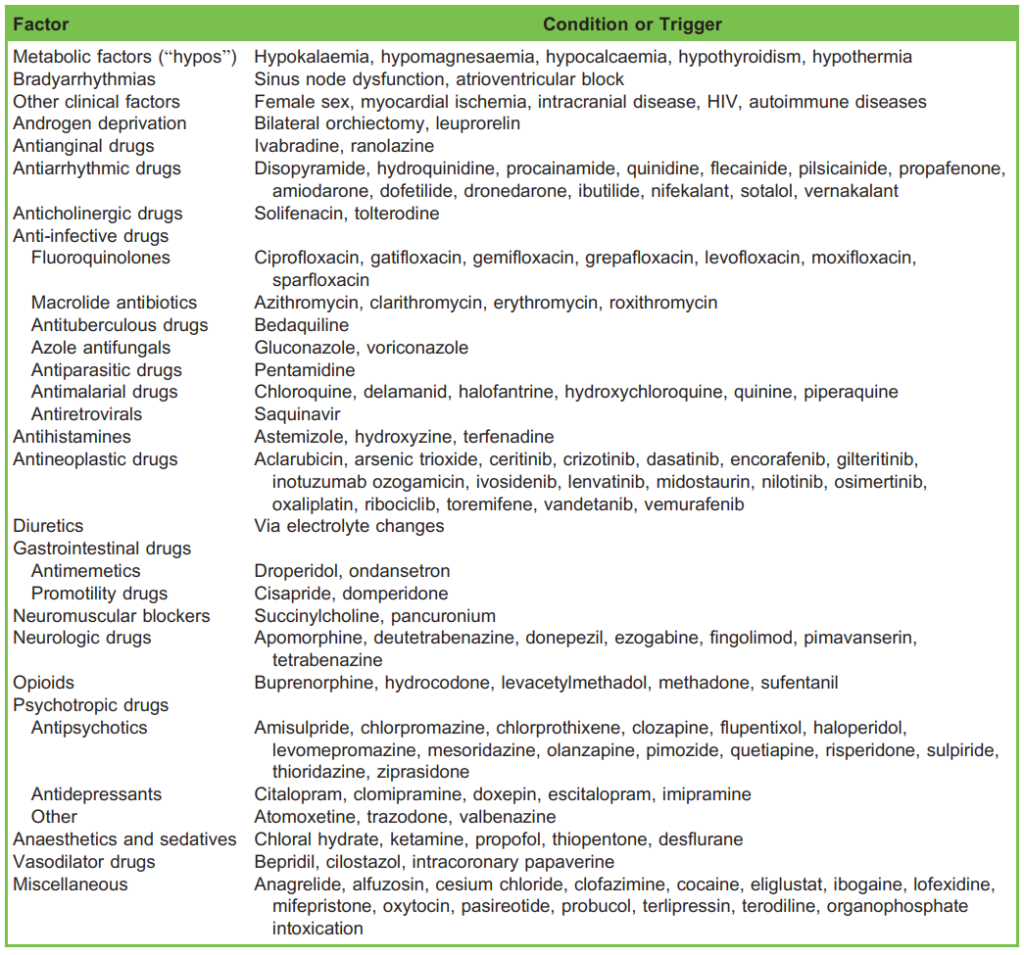
Table 1. Some Causes and Triggers of Long QT Syndrome (Both Congenital and Acquired)
DIAGNOSIS AND TREATMENT
Most individuals are asymptomatic, being diagnosed because of an affected family member or a prolonged QT interval that is detected by an ECG obtained for sports participation or anaesthesia. The primary symptoms, usually precipitated by a trigger
(Tables 1 and 2), include palpitations, arrhythmic syncope with or without generalised seizures and sudden cardiac arrest.12
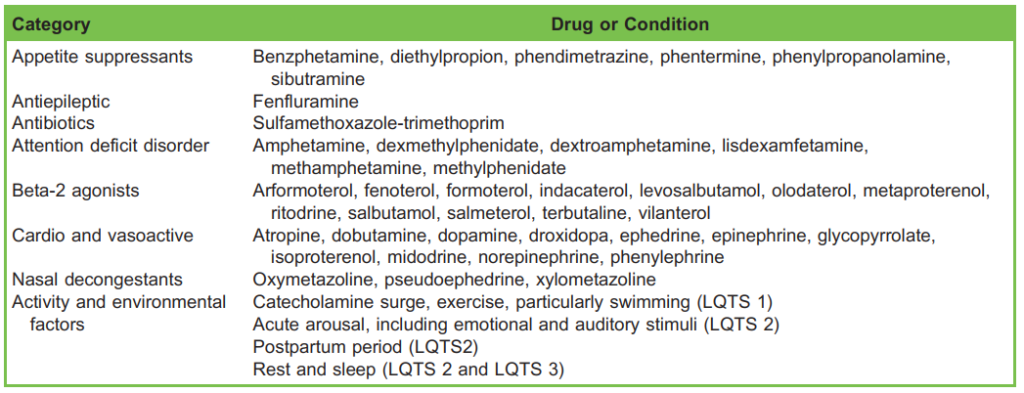
Table 2. Special Risk Factors in Congenital Long QT Syndrome (LQTS)
After obtaining a comprehensive personal and family history, as well as ECG monitoring, a Schwartz score should be calculated (Table 3) to determine the probability of the presence of congenital LQTS. Diagnosis of acquired LQTS can be made in a patient with sufficient QT prolongation that is associated with a known risk factor and that is reversible upon removal of the risk factor. If congenital LQTS is suspected, a cardiologic evaluation and genetic testing are advised.

Table 3. Schwartz Score: Diagnostic Criteria for Congenital Long QT Syndrome (LQTS). *The history of LQTS and unexplained sudden death should occur in 2 distinct family members to qualify for each set of points. †Mutually exclusive. ‡After excluding iatrogenic causes
Whilst most patients with congenital LQTS are started on a beta-adrenergic blocker, the cornerstone of the management of acquired LQTS is addressing the underlying cause. An implantable cardioverter defibrillator (ICD) should be considered for congenital forms in cases of extremely prolonged QTc or recurrent arrhythmias despite maximally tolerated doses of beta blocker.
PREOPERATIVE MANAGEMENT
The preanaesthetic evaluation should include review of personal or familial history of LQTS-related symptoms as well as all medications. Asymptomatic patients without a history of arrhythmic symptoms carry lower risk.
Preoperative 12-lead ECG is indicated to establish a baseline ECG. Cardiology consultation before elective surgery is warranted in cases with prior life-threatening arrhythmias, significant QT prolongation or anticipated interference with the normal function of cardiac implantable devices.
To prevent further QT prolongation and resulting arrhythmias, the following perioperative approach is warranted:
- Avoidance of QT prolonging drugs. The list is extensive and includes widely used drugs including antiarrhythmic, antibiotics, antihistamines, antidepressants, antipsychotics and antineoplastic agents (Table 1). An individual risk-benefit
analysis should be undertaken so that all nonessential treatments are paused. - Metabolic and electrolyte homeostasis. Hypokalaemia, hypomagnesaemia, hypocalcaemia and hypothyroidism further prolong repolarisation time. These imbalances should be identified and corrected prior to the surgery.
- Chronic beta-adrenergic blocker therapy when indicated. Beta-adrenergic blocking agents are the mainstay of treatment for congenital LQTS (class I recommendation in both the European and American Cardiological Societies’ guidelines). Propranolol is suggested given its superior efficacy, likely because of the additional inhibition of late sodium currents. This therapy inhibits both prolongation and dispersion of repolarisation, reducing the rate of malignant arrhythmias. It should be continued throughout the perioperative period (change to a parenteral formulation if nil by mouth). In contrast, there is no strong evidence to support chronic beta-adrenergic blockade in purely acquired LQTS.
INTRAOPERATIVE MANAGEMENT
The perioperative period is potentially life-threatening, since the patient is exposed to myriad risk factors, including haemodynamic instability, catecholamine surges, metabolic imbalances and exposure to drugs known to prolong the QT interval (although not necessarily torsadogenic).
Premedication and General Conditions
Benzodiazepines (in particular, midazolam) decrease the sympathetic tone and should be considered for anxiolysis, particularly in congenital LQTS. In addition, genotype-specific steps can be taken. For instance, loud noises may act as a trigger for those with the LQT2 genotype.6 Therefore, a calm environment in the operating theatre should be prioritised, including reduction of alarm sounds during induction and emergence.
Equipment and Monitors
The American Society of Anesthesiologists’ guidelines recommend standard monitoring comprising noninvasive arterial blood pressure, heart rate, continuous ECG (consider 5-lead), pulse oximetry, end-tidal carbon dioxide and body temperature. Before starting the case, it is important to check that a working defibrillator is located in theatre and that intravenous (IV) magnesium sulphate is available. In high-risk patients, the use of self-adhesive multifunction pads should be considered.
Overall Goals of Anaesthesia
Regardless of the surgery or anaesthesia technique chosen, maintenance of homeostasis is the priority. Avoid hypoxaemia, hypocapnia, hypercapnia and high-pressure ventilation. Sudden changes in heart rate and blood pressure should be anticipated and avoided. Maintain normothermia throughout the case and promptly correct any electrolyte disorders (especially hypokalaemia, hypomagnesemia and hypocalcaemia).
Induction and Maintenance of General Anaesthesia
Evidence suggests that propofol and opioids, such as fentanyl, alfentanil and remifentanil, are safe options for induction. In contrast, ketamine is not recommended because it may exacerbate haemodynamic adverse effects. A lidocaine bolus (1.5 mg/kg) before laryngoscopy may help to prevent catecholamine surges.
If muscle relaxants are required, rocuronium, vecuronium, atracurium and cisatracurium are safe. Succinylcholine and pancuronium may, however, cause undesired haemodynamic changes. Reversal agents with anticholinesterase activity should
be used with caution. Sugammadex used to reverse aminosteroid muscle relaxation is potentially a good option.
For maintenance of general anaesthesia, total IV anaesthesia with propofol is a good choice, although the QT prolongation by volatile anaesthetics (namely, isoflurane and sevoflurane) is probably clinically insignificant.
Regional Anaesthesia
Neuraxial and peripheral techniques are valid given their attenuation of the stress response and the effective analgesia they provide. However, adding epinephrine to the local anaesthetic solution is not advised. With regard to neuraxial anaesthesia,
epidural or combined spinal-epidural anaesthesia might allow better titration and, as such, is preferred over spinal anaesthesia because of the propensity for spinal anaesthesia to rapidly drop blood pressure. Consider sedation in awake patients.
Analgesia
Perioperative pain should be anticipated and adequately prevented and treated. Painful stimuli might induce a number of psychological and physiological responses, including enhanced sympathetic activity. In addition to regional techniques morphine, paracetamol and anti-inflammatory drugs are considered safe.
Antiemetics
Prophylactic antiemetic therapy is limited because of further prolongation of the QT interval by ondansetron and droperidol. As a result, dexamethasone, cyclizine and metoclopramide are preferred in this group of patients.
POSTOPERATIVE MANAGEMENT
All patients should be admitted to a postoperative care unit. Here, management should focus on the following:
- bed rest in a calm environment,
- continuous standard monitoring,
- adequate analgesia,
- control of postoperative nausea and vomiting and
- restarting chronic beta-adrenergic blockade in stable patients
MANAGEMENT OF CRITICAL ARRHYTHMIAS
Arrhythmias can occur at any time during the perioperative period. Prolongation and dispersion of repolarisation provide the substrate for early after depolarisations, triggered activity and reentry mechanisms. These events range from auto-limited episodes of TdP to sustained ventricular fibrillation.
TdP is a polymorphic ventricular tachycardia, with sinusoidal and cyclic alteration of the QRS axis, so that the peaks of the QRS complex seem to twist around the isoelectric line, as depicted in Figure 4. QT prolongation of the preceding beat is a typical feature and occasionally T-wave alternans (beat-to-beat variation in the shape or amplitude of T waves), a marker of cardiac electrical instability, may be seen.
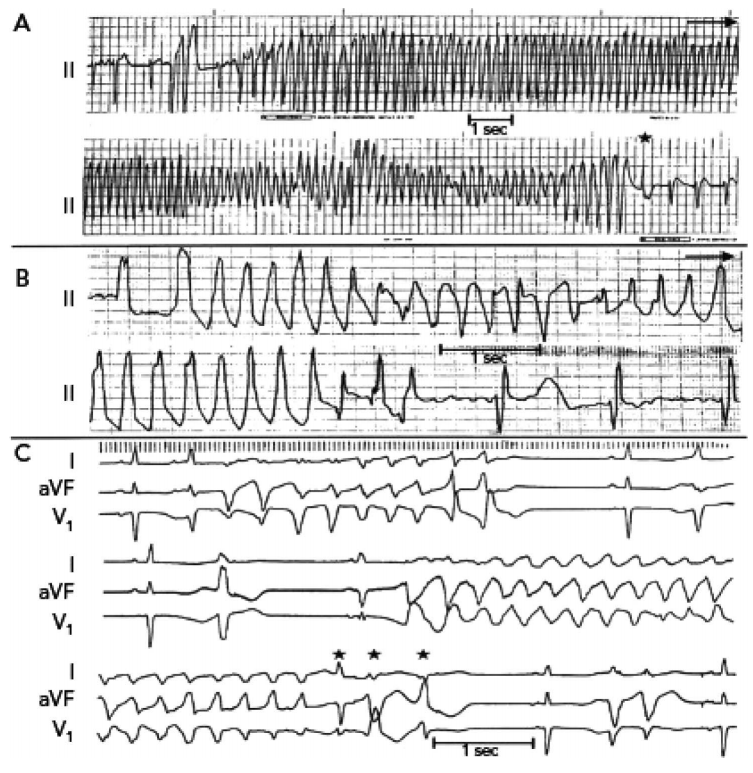
Figure 4. Three electrocardiograhic examples of long QT syndrome and torsades de pointes.15 Permission granted by Nabil E. El-Sherif, MD. This image/content is covered by the terms of the Creative Commons license (the CC-BY-NC 4.0) of this publication.
The torsadogenic potential of a drug is determined not only by prolongation of the QT interval but also by its ability to increase dispersion of repolarisation. This arrhythmia is usually short lived and terminates spontaneously; however, rapidly recurring episodes may degenerate into ventricular fibrillation and cardiac arrest. See Tables 4 and 5 for the prevention and treatment of LQTS and TdP.
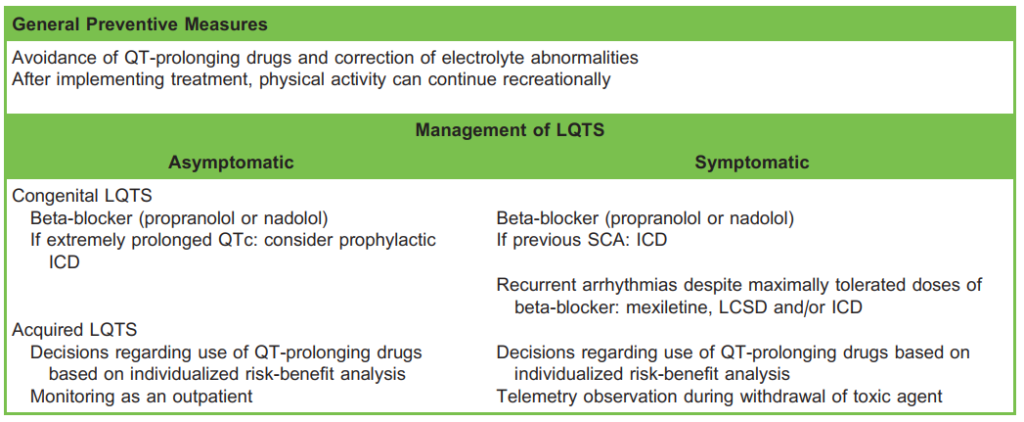
Table 4. Management of Patients With LQTS. LQTS indicates long QT syndrome; ICD, implantable cardioverter defibrillator; LCSD, left cardiac sympathetic denervation; SCA sudden cardiac arrest

Table 5. Acute Management of TDP
The management of various TdP arrhythmia scenarios is as follows:
- Single episode and haemodynamically stable: Regardless of magnesium levels, IV magnesium sulphate is the first-line therapy and helps to prevent recurrent arrhythmias. A single dose of 2 g or 8 mmol (4 mL of 50% magnesium sulphate mixed with dextrose 5% in water to a total volume of 50-100 mL) over 15 minutes is recommended. The mechanism is still unknown, but enhanced potassium efflux and inhibition of late calcium influx are suggested. Potential side effects of hypermagnesemia include enhanced neuromuscular blockade and respiratory depression as a result.
- Multiple self-terminating episodes and haemodynamically stable: Besides IV magnesium, consider temporary transvenous overdrive atrial pacing (set to 100-140 beats per minute) and/or IV isoproterenol (also known as isoprenaline) infusion: start at 2 μg/min and titrate to a heart rate of 100 beats per minute. Both strategies aim to reduce the RR interval and repolarisation time.
- Haemodynamic instability: Haemodynamically unstable patients due to ventricular arrhythmia require prompt synchronised electrical cardioversion, as well as 2 g IV magnesium.
- Pulseless arrhythmia: If TdP degenerates into ventricular fibrillation, commence cardiopulmonary resuscitation and attempt immediate defibrillation; 2 g IV magnesium should be administered. Avoid amiodarone as it has a proarrhythmic effect due the additional prolongation of the QTc interval. Give IV lidocaine instead.
Given the likelihood of arrhythmia recurrence, continuous monitoring (consider self-adhesive multifunction pads) and arrhythmia prevention and treatment are mainstay during the rest of the hospital stay.
SUMMARY
QT prolongation is a frequent phenomenon in the perioperative setting. Although most patients have an unremarkable course, some high-risk patients may develop malignant arrhythmias such as TdP. Table 6 summarises steps to be taken in the perioperative management to prevent these events.
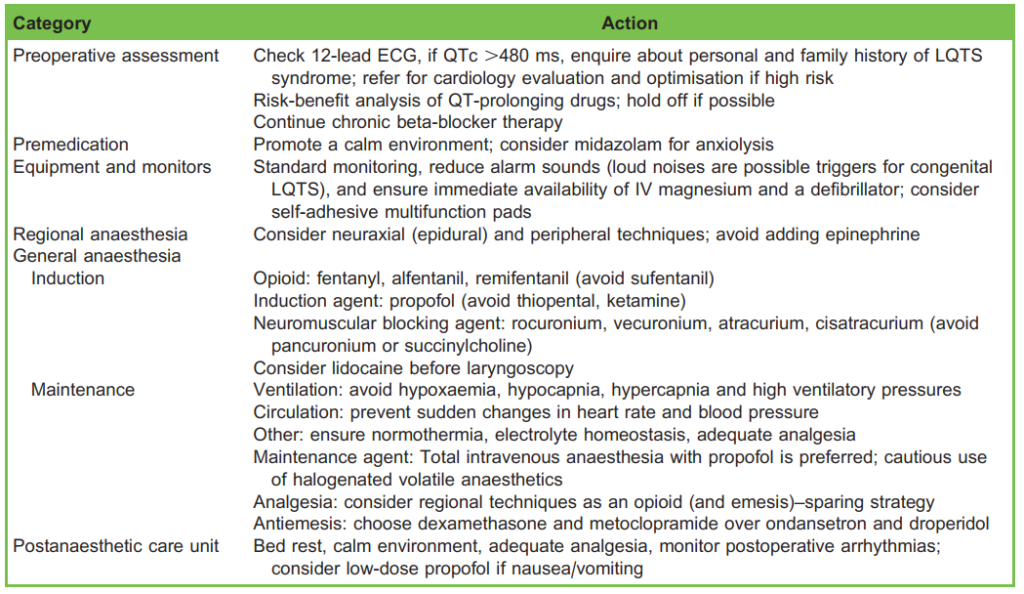
Table 6. Summary of Perioperative Management
REFERENCES
1. Schwartz P, Stramba-Badiale M, Crotti L et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761-17677.
2. Yu H, Zhang L, Liu J et al. Acquired long QT syndrome in hospitalized patients. Heart Rhythm. 2017;14:974-978.
3. Nagele P, Pal S, Brown F et al. Postoperative QT interval prolongation in patients undergoing noncardiac surgery under general anesthesia. Anesthesiology. 2012;117:321-328.
4. Bazett H. An analysis of the time-relations of electrocardiograms. Heart. 1920;7:353-370.
5. Vink A, Neumann B, Lieve K, et al. Determination and Interpretation of the QT interval. Comprehensive analysis of a large cohort of long QT syndrome patients and controls. Circulation. 2018;138:2345-2358.
6. Priori S, Blomstro¨ m-Lundqvist C, Mazzanti A, et al. 2015 ESC fines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2015;36:2793-2867.
7. Al-Khatib S, LaPointe N, Kramer JM, et al. What clinicians should know about the QT interval. JAMA. 2003;289:2120-2127.
8. Taggart N, Haglund C, Tester D et al. Diagnostic miscues in congenital long-QT syndrome. Circulation. 2007;115:2613-2620.
9. Schwartz P, Spazzolini C, Crotti L, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783-790.
10. Yang T, Roden D. Extracellular potassium modulation of drug block of IKr. Implications for torsade de pointes and reverse use-dependence. Circulation. 1996;93:407-411.
11. CredibleMedst. Search for Drugs that Prolong QT & induce Torsades de Pointes (TdP). Accessed on July 31, 2020. https://www.crediblemeds.org/index.php/drugsearch
12. Moss A, Schwartz P, Crampton R, et al. The long QT syndrome: prospective longitudinal study of 328 families. Circulation. 1991;84:1136-1144.
13. Ikonnikov G, Yelle D. Cardiac conducting system. Accessed November 8, 2020. http://www.pathophys.org/physiology-ofcardiac-conduction-and-contractility/
14. Giudicessi J, Ackerman M. Calcium revisited new insights into the molecular basis of long-QT syndrome. Circ Arrhythm Electrophysiol. 2016;9:e002480.
15. El-Sherif N, Turitto G, Boutjdir M. Acquired long QT syndrome and electrophysiology of torsade de pointes. Arrhythm Electrophysiol Rev. 2019;8:122-130
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/