Basic Sciences

Introduction
Disorders of sodium (Na+) and potassium (K+) are amongst the most common metabolic abnormalities seen by anaesthetists. They can be caused by a wide variety of pathological processes, and if untreated can very quickly become life threatening. Although the principles of assessment and treatment are simple, the basic physiology is often poorly understood. Mistakes in management are common, and can exacerbate the underlying problems.
Before reading this article, consider what you already know about Na+ and K+ by thinking about the following questions (the answers are within the text):
- What hormones control the way the kidney balances Na+ excretion and reabsorption?
- What factors influence the movement of K+ across the cell membrane?
- How can the causes of hyponatraemia be classified?
- How rapidly, and by what means, should dysnatraemia be corrected?
- Which methods of treatment of acute hyperkalaemia have been shown to be effective?
- In what ways are post-operative patients particularly at risk of electrolyte derangement?
This review initially revisits much of the important basic science involved in electrolyte balance with more clinical aspects discussed as the article progresses.
Distribution of sodium and potassium
The distribution of Na+ and K+ can be thought of as opposite – where one is found abundantly, the other is at low concentration. Sodium is the most prevalent cation in the extracellular fluid (ECF), with a normal level of around 140mmol/L, but has a typical intracellular concentration of around 10mmol/L. In contrast, potassium is the most prevalent cation in the intracellular fluid, with a concentration around 150mmol/L. Because the intracellular space is the largest fluid compartment in the body, this makes it the most abundant cation overall. Only around 1% of total body K+ is found in the plasma, and levels are kept between 3.5 and 4.5mmol/L.
The cell membrane acts as the barrier between the potassium-rich intracellular fluid and the sodium-rich extracellular fluid. While it allows free passage to water and to nonpolar, hydrophobic molecules, it is impermeable to large molecules or charged particles. Hence Na+ and K+ can only cross where specific carrier proteins allow them to do so.
In vivo, the membrane remains relatively impermeable to both Na+ and K+ most of the time. Excitable cells can change their permeability to allow the influx and efflux of ions that constitute an action potential. At rest, the large concentration gradients for Na+ and K+ are maintained by the action of Na+/ K+-ATPase, a transmembrane protein which pumps out 3 Na+ for each 2 K+ it pumps in. This also maintains the net negative resting membrane potential, since it involves a net transfer of one positive charge out of the cell on each cycle.
Although the Na+/ K+-ATPase maintains the concentration gradients across the cell membrane, other mechanisms are in overall control of total body Na+ and K+ levels.
Sodium homeostasis
The volume of circulating plasma is vitally important to the body, since an adequate plasma volume is required for normal tissue perfusion. The plasma volume is proportional to the ECF volume, and since Na+ is the major cation of the ECF, total body Na+ content is proportional to ECF volume.
In normal individuals, the kidney strives to achieve Na+ balance – that is, to have Na+ excretion equal to Na+ ingestion. The long-term control of blood pressure is achieved by the excretion or retention of Na+ (and hence plasma volume) in the kidney.
The vast majority (99-99.5%) of the Na+ that is filtered by the kidney is reabsorbed in the proximal tubule and the loop of Henle. This reabsorption seems to be largely fixed, even in sodium overload. There is much greater control over the 0.5% of filtered Na+ reabsorbed in the distal tubule and collecting ducts. It is this proportionately tiny amount, which allows the body to either retain sodium and water or excrete them when necessary. Various hormones influence this balance of retention and excretion.
-
Hormones increasing sodium reabsorption
- Renin:
- Released from the juxtaglomerular apparatus of the kidney
- Release is stimulated by: raised sympathetic tone, falling plasma volume, and certain prostaglandins, such as PGE2
- It has no direct effects promoting Na+ retention, although it controls the renin-angiotensin-aldosterone axis
- Angiotensin II:
- Levels rise as result of renin release
- In turn, it stimulates the release of aldosterone
- Also increases tone in the efferent glomerular arteriole. This leads to an increased filtration fraction, and hence a higher oncotic and lower hydrostatic pressure in the downstream, peritubular capillary. The net effect is to enhance Na+ reabsorption from the proximal tubule.
- Aldosterone:
- Steroid hormone released from the adrenal cortex
- End product of the renin-angiotensin-aldosterone system
- Acts on the distal tubule and collecting duct to increase Na+ and water reabsorption (proportionately more Na+ than water)
- Aldosterone release is also potentiated by hyperkalaemia
- Arginine vasopressin (AVP), also known as anti-diuretic hormone (ADH):
- Posterior pituitary peptide hormone, under direct control from the hypothalamus
- Two different receptor systems influence its release:
- Osmoreceptors in the hypothalamus itself sense changes in plasma osmolarity – ADH levels are either increased or decreased to keep osmolarity constant. A rising serum osmolarity is also the trigger for the thirst response, stimulating the drinking of water
- Baroreceptors in the carotid bodies sense changes in circulating volume – a fall in circulating volume causes a large increase in ADH concentration
- The stress response, as triggered by surgery, also causes ADH release
- Acts to cause passive absorption of water from the collecting ducts, concentrating the urine
- Also causes a small degree of Na+ reabsorption, but the retention of water is proportionately much greater
- Through its effects on total body water it can markedly effect the Na+ concentration
- In the absence of ADH activity (diabetes insipidus) there is an inability to concentrate the urine at all, with a resultant diuresis of up to 20L per day
- Renin:
-
Hormones increasing sodium excretion:
- Atrial Natriuretic Peptide (ANP):
- The main hormone opposing the above effects
- A small peptide produced from the atrial wall as a result of atrial stretching due to hypervolaemia
- Acts to increase Na (and hence water) excretion by increasing GFR and blocking Na reabsorption in the proximal collecting duct
- Some evidence suggests that other factors secreted by the hypothalamus, termed brain natriuretic peptides (BNP), may have similar roles.
- Atrial Natriuretic Peptide (ANP):
Potassium homeostasis
Small increases in the serum potassium concentration can be very quickly life threatening. The kidneys cannot excrete potassium quickly enough to contain surges due to oral potassium loads, and hence intracellular buffering plays an important role in homeostasis. As the kidneys excrete the excess and serum concentration falls, K+ is released again from the cells.
-
Factors enhancing potassium transport into cells:
-
- Insulin – via an increase in Na+ / K+ ATPase activity
- Adrenaline – via its action on beta-adrenoceptors
- Aldosterone – release stimulated by rising serum potassium levels
- Serum pH – as the pH falls, H+ enters the cells. If the rising H+ is due to accumulation of organic acid (e.g. lactate), the anion is able to permeate the cell along with its hydrogen ion. However, if there is accumulation of mineral acid (e.g. HCl) the inorganic ion will not cross the membrane. The cell must then excrete another cation to maintain electrical neutrality – and since K+ is most abundant, it is often exchanged. In the opposite situation, as pH rises the cells release H+ and in exchange take up potassium.
In the normal state of affairs, 90% of daily potassium intake is excreted via the kidneys and the rest via the colon. Around 90% of the filtered potassium load is reabsorbed by the start of the distal tubule, and this figure is largely constant in a wide range of potassium intake. The overall urinary excretion of K+ is therefore controlled by the distal tubule and collecting ducts.
In these parts of the kidney, reabsorption of Na+ through specialised channels provides a substrate for Na+/ K+-ATPase on the basolateral cell surface, and hence movement of K+ from the peritubular fluid into the lumen. This is enhanced by a negative electrical gradient in the intraluminal fluid (since Na+ is reabsorbed without its anion). In situations of potassium depletion, a K+/ H+ -ATPase on the luminal membrane exchanges K+ for hydrogen ions, helping to explain the metabolic alkalosis often encountered in potassium deficiency.
-
Factors influencing renal potassium handling:
-
- Aldosterone – enhances activity of Na+/ K+-ATPase in the distal tubule and collecting duct; secretion is directly stimulated by high potassium levels
- Glucocorticoids – act on the same renal components as aldosterone; usually metabolized by renal 11-beta-hydroxysteroid dehydrogenase, but in glucocorticoid excess this enzyme can be overwhelmed, which is the cause of hypokalaemia seen in steroid treatment or Cushing’s Syndrome
- Increased tubular flow rate – (seen with volume expansion) the increased flow “washes out” or dilutes the K+ excreted into the tubule, favouring further K+ excretion down a concentration gradient
- Extracellular pH – K+ is exchanged for H+ in the tubular fluid; alkalosis enhances H+ reabsorption, and hence Na+ excretion, and acidosis has the opposite effect.
- Diuretics – enhance flow rate to the distal tubule, increasing potassium washout (as above). Loop diuretics (furosemide) also reduce K+ reabsorption from the thick ascending limb of the loop of Henle. Potassium-sparing diuretics (amiloride, triamterene) block Na+ reabsorption in the late distal tubule and collecting duct. Because this Na+ influx is what creates the negative electrical gradient in the tubule fluid driving K+ excretion, they lead to a decrease in potassium excretion. Spironolactone is an aldosterone antagonist and hence blocks Na+ reabsorption and K+ excretion in the collecting ducts.
Causes of hyponatraemia
Consideration of the osmotic state of the patient is essential in the evaluation of hyponatraemia:
- Normal Osmolarity: Pseudohyponatraemia
- Due to a measurement error which can result when the solid phase of plasma (that due to lipid and protein) is increased
- Typically caused by hypertriglyceridaemia or paraproteinaemia.
- High Osmolarity: Translocational hyponatraemia
- Occurs when an osmotically active solute that cannot cross the cell membrane is present in the plasma.
- Most solutes such as urea or ethanol can enter the cells, and cause hypertonicity without cell dehydration.
- However, in the case of the insulinopaenic diabetic patient, glucose cannot enter cells and hence water is displaced across the cell membrane, dehydrating the cells and “diluting” the sodium in the serum.
- This is also the cause of hyponatraemia seen in the TURP syndrome, in which glycine is inadvertently infused to the same effect.
- Low Osmolarity: True hyponatraemia
- True hyponatraemia is always a hypo-osmolar condition
- The next stage is to consider the volume status of the patient:
- Hypovolaemic hyponatraemia:
- Loss of both sodium and water, but proportionately more sodium
- Caused by solute and water losses from either a renal or gastrointestinal source
- Usually these patients are consuming water or hypotonic fluid, although not in quantities sufficient to restore normovolaemia
- An estimation of the urinary sodium level can be helpful: a level below 30mmol/L suggests an extrarenal cause, while a level above 30mmol/L suggest a primary renal problem.
- Euvolaemic hyponatraemia:
- The most common form seen in hospitalized patients.
- May have a slight increase or decrease in volume, but it is not clinically evident, and they do not have oedema.
- The most common cause is the inappropriate administration of hypotonic fluid.
- The syndrome of inappropriate ADH secretion (SIADH) also causes euvolaemic hyponatraemia; in order to make this diagnosis one must first exclude renal, pituitary, adrenal or thyroid dysfunction, and the patient must not be taking diuretics.
- Hypervolaemic hyponatraemia:
- Characterised by both sodium and water retention, with proportionately more water.
- Therefore have an increased amount of total body sodium.
- Causes are all characterised by disordered water excretion, and are usually easy to diagnose.
- Hypovolaemic hyponatraemia:
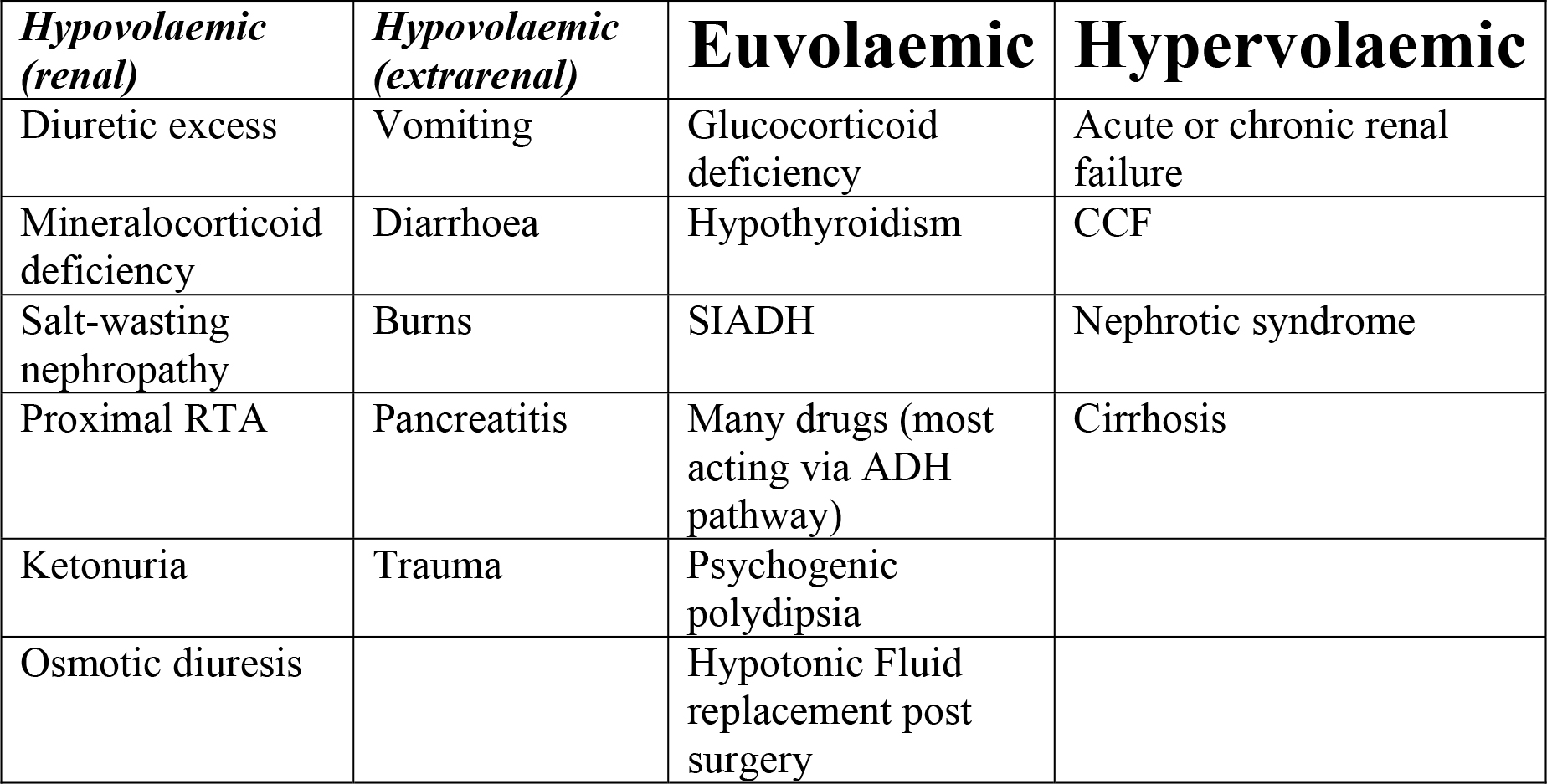
Figure 1 Causes of hyponatraemia
Causes of hypernatraemia
Hypernatraemia is either caused by excessive salt intake, or (much more frequently) inadequate water intake. As with hyponatraemia, consideration of the volume status of the patient is essential:
- Hypovolaemic hypernatraemia:
- Loss of both sodium and water, but relatively more water
- An estimation of the urinary sodium level can be helpful: a level below 30mmol/L suggests an extrarenal cause, while a level above 30mmol/L suggest a primary renal problem.
- These patients are either not able to take in adequate fluid to replace their losses, or are prevented from doing so.
- Euvolaemic hypernatraemia:
- Occurs when body water losses are partially replaced.
- May be due to lack of availability of water, or due to a blunting of the normal thirst response seen in the extremes of age.
- Hypervolaemic hypernatraemia:
- Seen where sodium retention is not matched by increased fluid intake.
- More uncommon than the other two types of hypernatraemia.
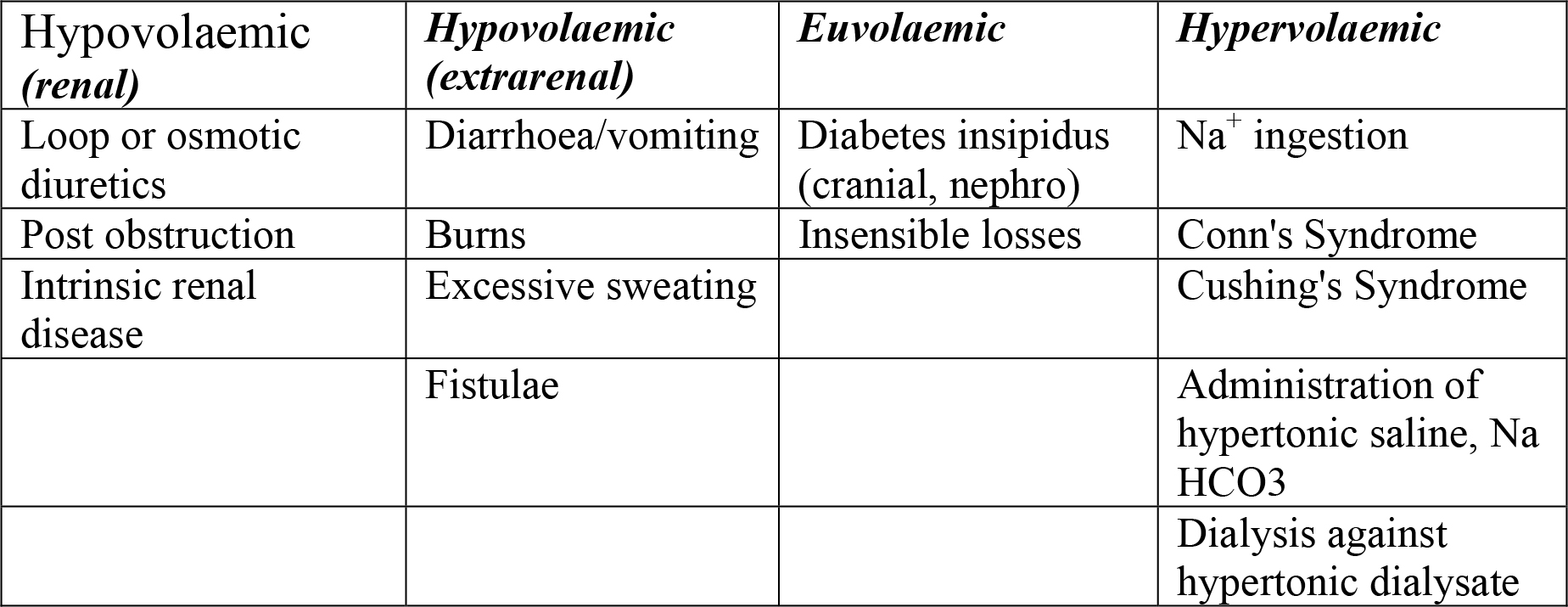
Figure 2 Causes of hypernatraemia
Causes of hypokalaemia
Hypokalaemia is caused by a shift of potassium into cells, or more commonly by a total body potassium deficit. Occasionally the two situations may co-exist.
- Intracellular potassium shifting:
- Caused by:
- excess insulin (exogenous or endogenous)
- beta-adrenoceptor agonists (such as endogenous catecholamines or exogenous salbutamol)
- theophylline toxicity
- acute rise in plasma pH.
- Caused by:
- Total body potassium deficit:
- May result from either decreased intake or increased losses.
- Diet must be severely deficient in K+ over a long period in order to reach a position of clinical hypokalaemia; hence seen most commonly in alcoholics.
- Excessive losses may be either renal or extrarenal:
- Renal causes include:
- Diuretics
- Mineralocorticoid excess
- Glucocorticoid excess
- Renal tubular acidosis Type I and II
- DKA – glucose causes an osmotic diuresis, washing out potassium
- Vomiting – this is not caused by a loss of K+ in the vomit; rather, loss of H+ and water lead to metabolic alkalosis and increased aldosterone
- Ureterosigmoidostomy
- Rare inherited conditions such as Bartter’s and Gitelman’s Syndromes
- Extrarenal causes include:
- Inadequate intake
- Excessive perspiration
- Chronic diarrhoea
- Gastrointestinal fistulae
- Renal causes include:
Causes of hyperkalaemia
Hyperkalaemia may be due to either an overall increase in total body potassium, or an acute shift of potassium from the intracellular to the extracellular compartment.
- Extracellular Potassium Shifts:
- Caused by:
- Acidosis – H+ is taken into the cell in exchange for K+
- Insulin deficiency, with hyperglycaemia – note that this is often found coexistent with a profound total body potassium deficit
- Digitalis toxicity – due to inhibition of the Na+/ K+ ATPase
- Beta-blockers – typically cause only a mild elevation in K+
- Exercise – Potassium efflux from skeletal muscle as a result of muscular contraction
- Suxamethonium administration
- Fasciculations lead to an efflux of potassium from skeletal muscle, similar to the effect of exercise but more pronounced and more acute.
- A single 100mg dose may cause the serum potassium to rise by up to 1.0mmol/L. If the patient already has elevated serum potassium, this may be enough to cause a fatal arrhythmia.
- In patients with denervated muscle, the usual mechanisms keeping the acetylcholine receptors in the synaptic cleft are disturbed, and they spread out to cover the whole of the muscle fibre (extrajunctional receptors).
- Suxamethonium administration is contraindicated in these patients as it causes a much bigger potassium efflux and often leads to dangerous hyperkalaemia.
- Caused by:
- Excessive potassium input:
- Caused by:
- Cellular lysis, as in haemolysis or rhabdomyolysis
- Inappropriate prescription of K+ containing IV fluids or supplements is a very important cause in hospitalized patients.
- Caused by:
- Impaired renal excretion:
- Caused by:
- Decreased GFR – renal failure is the commonest cause of hyperkalaemia
- Mineralocorticoid insufficiency – this may be due to primary adrenal failure, hyporeninaemic hypoaldosteronism (RTA Type IV), or due to drugs like ACE inhibitors, ATII-receptor antagonists or spironolactone
- Potassium sparing diuretics – see above
- Primary renal insults (such as interstitial nephritis) causing decreased potassium excretion in the distal tubules and collecting ducts
- Caused by:
- Pseudohyperkalaemia:
- A common cause of spuriously elevated potassium levels.
- The most common causes are in vitro haemolysis, or leaving the tourniquet on for an extended period prior to blood sampling.
- It is also seen in patients with highly elevated white cell or platelet counts, due to secretion of potassium from these cells prior to laboratory analysis.
Implications for clinical anaesthesia
In general, the implications of an increase or decrease in the serum levels of Na+ or K+ are dependent on the speed with which this change occurred. Very low levels of Na+ can be reached without appreciable symptoms if they have come about gradually over several months. Similarly, patients with chronic renal impairment can often tolerate levels of hyperkalaemia that would be fatal if they happened over a few hours. However, there seem to be limits above or below which the normal physiological processes are affected no matter how long the system takes to get there.
Patients can also be at risk from overly rapid correction of electrolyte imbalance. In deciding when and how to treat a patient, a balance must be struck between the risks of the condition, and the risks of treatment.
Effects of hyponatraemia and its treatment
The normal range of serum sodium is usually quoted as being approx. 135-145mmol/L; however, levels between 125mmol/L and 150mmol/L are often asymptomatic. Outside this range there is an increasing frequency of nausea, lethargy, weakness and confusion, and levels above 160mmol/L or below 110mmol/L are strongly associated with seizures, coma and death.
As serum sodium and osmolarity fall, water tends to enter the cells causing them to swell. Clinically this is most important in the brain.
Several factors put patients at increased risk of complications of hyponatraemia or its treatment:
- Post-operative patients, premenopausal women, elderly women taking thiazides, children, and patients who are hypoxaemic are all at increased risk of acute hyponatraemic cerebral oedema.
- Malnourished patients, alcoholics, those with burns or hypokalaemia are all at increased risk of osmotic myelinolysis due to overly rapid correction of hyponatraemia.
A recent review of the literature has pointed out that there is as yet no consensus on the optimum treatment of dysnatraemia. However, all authorities stress the importance of distinguishing between hyponatraemia that has developed acutely (usually taken to mean over <48hrs) and chronic hyponatraemia. This is because of important differences in the management between the two groups.
Most authors suggest that hyponatraemia that has developed acutely (for instance, in the immediate post-operative period) can be safely treated with rapid correction. Rapid correction should only be undertaken in patients who are symptomatic, and the aim of treatment is to correct the level until the symptoms resolve. Some sources have suggested that correction by up to 2mmol/L/hr is safe in the initial treatment of acute hyponatraemic states. Correction to a serum [Na+] of >135mmol/L may be safe in this situation, but it is not necessary to correct rapidly once the symptoms have resolved.
Methods of rapid correction might include the administration of furosemide and/or hypertonic saline, but in this case management should be by a specialist in an appropriate setting, with monitoring of serum Na+ levels hourly.
The treatment of chronic hyponatraemia is also determined by the presence or absence of symptoms. In the presence of symptoms, a rapid correction of up to 10mmol/L may be permissible. Following this, however, the rate of reversal should be limited to 1.5mmol/L/hr, and to no more than 8mmol/L over 24hrs. Some sources suggest that a rate of 12mmol/L in 24hrs is safe.
Fluid restriction is the mainstay of treatment in these patients, who need to have regular neurological assessment and rechecking of serum electrolytes at least every 12hrs. In the long-term, treatment is aimed at identifying and dealing with the underlying cause. Future advances in the shape of selective ADH (AVP) antagonists (so-called aquaretics) look set to improve the long-term management of chronic hyponatraemia.
In all cases, hypovolaemia if present must be corrected first with 0.9% saline. This removes the ADH response that is accentuating the sodium/water imbalance. In patients who are hypervolaemic, the treatment is aimed at fluid restriction, salt restriction and loop diuretics. Aquaretics may also be useful drugs for these patients as well.
While evidence is lacking that chronic hyponatraemia is associated with worse surgical outcomes, anything more than mild, asymptomatic hyponatraemia should be regarded as a relative contraindication to elective surgery.
Treatment of hypernatraemia
Firstly any volume deficit should be corrected with 0.9% saline until the hypovolaemia, as measured by orthostatic hypotension, improves. The cause of fluid loss should also be investigated and treated.
The total body water deficit can be calculated based on the serum sodium and the assumption that 60% of the body is water – this deficit should then be corrected with 5% dextrose, with half given in the first 12-24hrs, and the rest over the next 24-36hrs. In the case of hypervolaemic hypernatraemia, the removal of excess sodium is the aim, and loop diuretics or dialysis may achieve this if the patient has renal dysfunction.
Effects of hypokalaemia
The effects of hypokalaemia depend upon the serum level. A normal value of 3.54.5mmol/L is generally accepted, but levels of 3.0-3.5mmol/L are usually asymptomatic. Below 3.0mmol/L general symptoms of weakness, lassitude and constipation are common. Below 2.5mmol/L muscle necrosis has been described (probably due to an impaired ability to increase blood flow during exercise), and below 2.0mmol/L an ascending paralysis may be seen, eventually leading to respiratory compromise.
Patients without underlying cardiac disease are unlikely to suffer myocardial effects, even at levels below 3.0mmol/L. However, those with ischaemic heart disease, heart failure or left ventricular dysfunction are at risk of arrhythmias with only mild or moderate hypokalaemia. Initially U-waves are seen on the ECG, with gradual sagging of the ST segment and flattening of the T-wave. Slight widening of the QRS complex and PR elongation may be seen, and there is a predisposition to both ventricular and supraventricular ectopic rhythms, especially in a patient taking digoxin.
Renal effects of hypokalaemia include metabolic acidosis, increased ammoniagenesis and numerous structural changes in the kidney if the condition persists.
As with sodium, the rapidity of the change in K+ level has a large influence on the severity of the symptoms.
Treatment of hypokalaemia
Once intracellular K+ shifts have been excluded (theophylline toxicity, hyperinsulinaemia) the treatment of hypokalaemia is aimed at replacement of potassium. Ideally, this should be oral supplementation, but if severe the initial replacement is best given intravenously, through a central vein in the context of a critical care facility. Careless administration of intravenous potassium is the commonest cause of hyperkalaemia in hospitalized patients, so appropriate consideration should be given to this decision. In any case, the rate of administration should not exceed 20mmol/hr, and the patient should have continuous cardiac monitoring.
In the absence of factors causing potassium shifting into cells, the serum potassium is a good guide to the total body potassium deficit. A fall from 3.5 to 3.0mmol/L suggests an approximately 5% deficit in total stores (around 175mmol); a decline from 3.0 to 2.0mmol/L suggests a further 200-400mmol deficit. Magnesium deficiency is very commonly associated with hypokalaemia and levels should be checked and magnesium replaced if appropriate.
Prophylactic administration of potassium to post-operative patients at risk of cardiac abnormalities is a common practice. There is evidence that minor elevation of K (within the normal range) can reduce the incidence of electrocardiac abnormalities such as Uwaves, bifid t-waves and signs of digitalis toxicity. There is some evidence of benefit in maintaining K levels between 4.0-4.5 in patients post cardiac surgery, and in patients on drugs such as quinidine and sotalol (which potentially predispose to Torsades de Pointes). Additionally, potassium supplementation may benefit patients with abnormal repolarisation in the context of congestive cardiac failure. However, the practice of artificially augmenting the potassium level to abolish single ventricular ectopic beats or as a routine treatment for all post-operative patients is no longer considered best practice.
Effects of hyperkalaemia
The most important effects of hyperkalaemia are on the heart. Levels below 6.0mmol/L rarely cause any clinical symptoms. As the serum K+ level increases, ECG changes are noted: firstly peaking of the T-waves, then broadening of the P-waves and QRS-complex when the level is >7.0mmol/L. Finally the ECG takes on a sinusoidal pattern, which is a precursor to cardiac arrest. Terminal ECG changes may develop very quickly, and even with mildly elevated potassium levels any sign of ECG involvement should prompt immediate treatment.
As with other electrolyte disturbances, the speed of onset of hyperkalaemia is very important. A relatively small increase, if it occurs over a short time, can precipitate a fatal arrhythmia where a much higher level may be tolerated (for instance, in the insidious onset of renal failure) if it has developed over a longer period.
Other sequelae of hyperkalaemia include parasthesiae, weakness, paralysis, a decreased renal production of ammonia, an increased renal retention of H+ and a subsequent metabolic acidosis, natriuresis, and elevated levels of aldosterone and insulin.
Treatment of hyperkalaemia
The various therapies commonly used to reduce the K+ level acutely may be divided into two groups: those that seek to transiently move the potassium to the intracellular compartment, and those that seek to remove an overall surplus of potassium from the body. While the former group may be used in the vast majority of hyperkalaemic patients, not all hyperkalaemic patients have excess total body potassium.
The classic example is an acidotic patient with diabetic ketoacidosis, who has an elevated serum potassium due to cellular impermeability in the absence of insulin, but who is often profoundly depleted in potassium levels overall. Such patients require emergency management to lower the high potassium levels they present with, but as treatment commences, and their cells rapidly become permeable to potassium, caution must be taken to avoid them developing a rebound hypokalaemia.
A recent Cochrane review of the efficacy of various potassium lowering therapies showed that, despite this being an exceedingly common problem in hospitalized patients, very little evidence exists to guide the practitioner towards the most effective. The therapies most often suggested for acute potassium lowering are infusions of glucose and insulin, beta2-adrenoceptor agonists, either nebulized or inhaled, and IV sodium bicarbonate. Of these, glucose-insulin and beta-agonists both seem to be effective, and a combination seems more effective than either being used in isolation. The evidence for sodium bicarbonate is equivocal.
The same review investigated two methods for removing excess potassium from the system: K+-absorbing styramine resins, and dialysis. Of these, the evidence was that resins were not effective at 4 hrs post administration, but longer-term studies have not been done. Dialysis was effective at decreasing total body potassium over the same period.
In addition, the administration of calcium (as either calcium gluconate or calcium chloride) is recommended as a means of rapidly reversing the repolarisation abnormalities seen in severe hyperkalaemia. Its use is supported by experimental and animal studies, but there are neither randomised trials nor any good evidence to recommend one formulation over another. Furthermore, it must always be remembered that a cornerstone of treatment is to diagnose the underlying cause of hyperkalaemia and take steps to reverse this.
Conclusions
Disorders of Na+ and K+ homeostasis are very common problems, encountered in clinical practice on an almost daily basis. They are frequently mismanaged due to poor understanding of Na+ and K+ metabolism. Careless prescription of perioperative fluids and infrequent checking of electrolyte levels puts patients needlessly at risk. Often it is the most junior doctors involved in a patient’s care who have responsibility for these areas.
Surgical patients are frequently affected by electrolyte imbalance. They are often sedated or not allowed to eat and drink, and hence have intravenous fluid infusions prescribed for extended periods. Pre-operative bowel obstruction or bowel prep can leave them profoundly dehydrated. They are subject to large fluid shifts in theatre, and postoperatively are usually in a water-retaining state due to a stress response and ADH secretion.
Further reading
- Reynolds RM, Padfield PL, Seckl JR. Disorders of sodium balance. BMJ 2006; 332: 702-5
- Mahoney BA, Smith WAD, Lo DS, Tsoi K, Tonelli M, Clase CM. Emergency interventions for hyperkalaemia. The Cochrane Database of Systematic Reviews. 2006 Issue 2
- Gennari FJ. Current Concepts: Hypokalemia. N Engl J Med. 1998; 339:451-458
- Pinski SL. Potassium replacement after cardiac surgery: It is not time to change practice, yet. Crit Care Med. 27(11): 2581-2582
- Schrier RW. Atlas of Diseases of the Kidney, Volume I, Chapters 1-3. Blackwell Science, 1999
- Adrogué HJ, Madias NE. Primary Care: Hyponatremia. N Engl J Med. 2000; 342:1581-1589
- Adrogué HJ, Madias NE. Primary Care: Hypernatremia. N Engl J Med. 2000; 342:1493-1499
- Sterns RH, Riggs JE, Schochet SS. Osmotic demyelination syndrome following correction of hyponatremia. N Engl J Med. 1986; 314:1535-1542
- Decaux G, Soupart A. Treatment of symptomatic hyponatremia. Am J Med Sci. 2003; 326(1): 25-30
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/