Basic Sciences

QUESTIONS
Before continuing, try to answer the following questions. The answers can be found at the end of the article.
- The anatomical features of Tetralogy of Fallot may include
- Ventricular septal defect
- Coarctation of the aorta
- Aberrant pulmonary venous drainage
- Abnormal coronary artery anatomy
- Right ventricular hypertrophy
- Treatment prior to full correction may include
- Pulmonary artery banding
- Beta blocking drugs
- A prostaglandin infusion
- Diuretics
- Features of a hypercyanotic spell are
- Irritability
- Back arching
- 2:1 heart block
- Cyanosis
- Death
- Management of a Tetralogy ‘spell’ may include
- Sedation with morphine
- Limiting inspired oxygen
- Increasing inspired oxygen
- Sodium nitroprusside
- Phenylephrine
WHAT IS TETRALOGY OF FALLOT?
Anatomy and variants
Described by the French physician Etienne-Louis Arthur Fallot in 1888, Tetralogy of Fallot (TOF) consists of four anatomical components (see Fig 1):
- Ventricular septal defect
- An abnormally positioned aortic valve above (overrides) the ventricular septum
- Right ventricular outflow tract (RVOT) obstruction.
- Right ventricular myocardial hypertrophy.
There are four major variants of TOF:
- TOF with pulmonary stenosis, either subvalvar, valvar or supravalvar, or any combination of the three. This is the most common variant and is the focus of this tutorial.
- TOF with pulmonary atresia. This is a severe variant commonly associated with hypoplastic pulmonary arteries with or without collaterals between the aorta and the pulmonary arteries (major aortopulmonary collateral arteries, MAPCAs).
- TOF with absent pulmonary valve syndrome is uncommon. The pulmonary valve is dysplastic and the pulmonary arteries dilated causing bronchial compression, which may result in breathing difficulties.
- TOF with atrioventricular septal defect. This is the least common variant, and complete repair is challenging from a surgical perspective.
Coronary artery anatomy is abnormal in 10% of cases; in the most severe cases the right coronary artery crosses the RVOT. This affects the options for surgical repair.
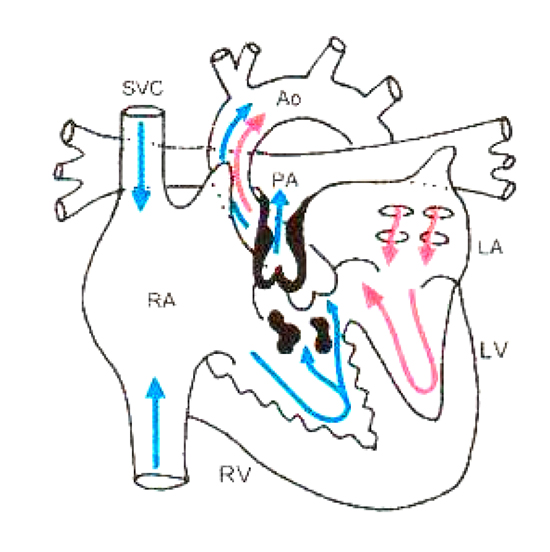
Figure 1. Tetralogy of Fallot with subvalvar, valvar and supravalvar pulmonary stenosis. Reproduced with kind permission from: Pediatric Heart Surgery (A Ready Reference for Professionals). L Eliot May. http://www.maxishare.com/
PATHOPHYSIOLOGY AND NATURAL HISTORY
Tetralogy of Fallot is the most common form of cyanotic heart disease and accounts for 3.5% of all cases of congenital heart disease (1:3600 live births in the UK). The cause of TOF is unknown, but 25% of patients have a chromosomal abnormality (microdeletion at 11q position on chromosome 22), which may be associated with immune deficiency (Di George syndrome), or velocardiofacial syndrome and submucous cleft palate.
The clinical features of TOF are due to reduced pulmonary blood flow and cyanosis and are variable in severity, depending on the degree of obstruction at the RVOT. Obstruction causes deoxygenated blood to shunt from the right ventricle to left across the usually large (unrestrictive) VSD. A harsh ejection systolic murmur is present, and is due to turbulent flow through the RVOT.
In most babies, pulmonary blood flow is adequate in the first few weeks of life, but the child becomes increasingly cyanosed with age. Some babies have severely compromised pulmonary blood flow from birth, or in the case of pulmonary atresia without significant pulmonary collateral vessels, the baby will have ‘duct dependent’ pulmonary circulation. These babies will require an early palliative pulmonary shunt (mBT shunt). Some children have minimal right ventricular outflow obstruction (RVOTO), and the history is more typical of a VSD with minimal cyanosis (‘pink tet’).
Some infants may have dynamic obstruction at the pulmonary infundibulum (subvalvar RVOTO). This leads to ‘hypercyanotic spells’, which are a classical symptom of TOF. The child becomes deeply cyanosed after an episode of crying and may become limp and unresponsive. This is a sign of acute reduction in pulmonary blood flow associated with sudden increase in the dynamic obstruction to the right ventricular outflow tract. It occurs due to muscular spasm at the subpulmonary infundibulum secondary to elevated levels of endogenous adrenaline. During a spell the cyanosis may be relieved if the baby is placed in the knee to chest position. Such babies respond well to a β-blocker such as propranolol before definitive surgery is carried out.
Older children with uncorrected TOF will develop severe clubbing (see Fig 2), and may learn to ‘squat’ to increase pulmonary blood flow – this position increases the systemic vascular resistance and reduces the right-to-left shunt across the VSD (see Fig 3). They may be polycythaemic and suffer complications of long standing polycythaemia:
- Coagulopathy
- Intracranial abscess
- Stroke
- Hyperuricemia
- Neurodevelopmental delay
Adolescents and adults will have reduced exercise tolerance and are vulnerable to ventricular arrhythmias.
Without surgical intervention, around 50% of children with TOF will die within the first few years of life and survival beyond 30 years without surgery is uncommon. With corrective surgery in childhood, survival to discharge from hospital is 95-99%, and almost all children can be expected to survive to adulthood.
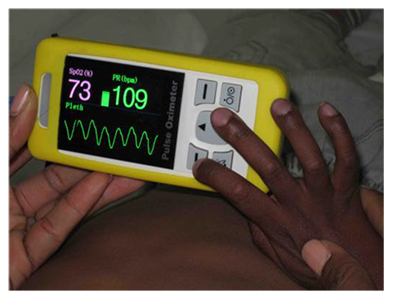
Figure 2. Finger clubbing in a child with TOF

Figure 3. A child with TOF ‘squatting’
DIAGNOSIS
History
The diagnosis may be made antenatally by ultrasound scan at 20 weeks. If this is the case the mother should be referred to a paediatric cardiologist for counselling and so that the child can be assessed at birth, particularly if the child may have duct dependent pulmonary circulation. The diagnosis may be made postnatally with the observation of cyanosis or a murmur. Later, the child will become increasingly cyanosed and may develop cyanotic spells.
Case history
A 10 month old presented for complete repair of Tetralogy of Fallot. She had been diagnosed at birth due to the presence of cyanosis. She developed symptomatic cyanotic ‘spells’ at two months of age, which occurred after feeding or when she became upset. The parents managed these by trying to settle her, but as the spells became more frequent, treatment with oral propranolol was started. She remained cyanosed with only occasional spells. Elective surgery for complete repair was delayed to 10 months of age due to the presence of an aberrant branch of the right coronary artery crossing the right ventricular outflow tract. Preoperative oxygen saturations were noted to be approximately 60% in air. Midazolam was given as an oral premed. Anaesthesia was induced with sevoflurane in oxygen – a peripheral cannula, arterial line and 5Fr 5cm internal jugular line were sited. During the pre-bypass period, the child has episodes of severe desaturation to 60%, despite a FiO2 of 1.0. Treatment of the hypercyanotic spell was with Hartmann’s solution 20 ml.kg-1, boluses of phenylephrine1mcg.kg-1, followed by an infusion of noradrenaline at 0.1mcg.kg-1.min-1. Complete surgical correction was achieved on cardiopulmonary bypass. Separation from bypass was achieved after loading with milrinone 50mcg.kg-1, continued as a maintenance rate of 0.5 mcg.kg.min-1 with noradrenaline 0.02 mcg.kg-1.min-1. The remainder of the postoperative course was uneventful; the child was extubated the following day, inotropic support was withdrawn over 24 hours, and the child was discharged from hospital on day 6.
Examination
Typical signs in TOF include:
- Central cyanosis. This may be missed in a child with severe anaemia or children with pigmented skin. It is detected by looking at the colour of the tongue – blue colouration suggests a saturation of less than 85%. Central cyanosis is not improved by breathing 100% oxygen and its presence should be confirmed by a pulse oximeter.
- Clubbing, seen from 3-6 months of age
- Long harsh ejection systolic murmur heard best over the pulmonary area and the left sternal border. A thrill may be felt along the left sternal border.
In older children with unrepaired TOF look for signs suggestive of complications of long-standing cyanosis:
- Stroke, intracerebral abscess
- Neurodevelopmental delay
- Arrhythmias
Associations
The presence of TOF should raise the suspicion of associated anomalies. These include:
- Di George syndrome (22q11 deletion). This is typically associated with learning difficulties, cleft palate, hypocalcaemia, absent thymus (frequent respiratory infections) and a typical facial appearance. Patients with Di George syndrome will require irradiated blood for transfusion as they have absent T cells and immunocompetent white cells in donor blood may result in ‘graft versus host’ disease.
- Isolated cleft lip and palate
- Hypospadias
- Skeletal abnormalities.
Special Investigations
Oxygen saturation
- Typical SpO2 65%-85%
Haematology
- Full blood count – haemoglobin will be elevated in proportion to the degree of cyanosis.
- Coagulation studies – severe polycythaemia may result in reduced coagulation factors and reduced platelet count.
Chest X-ray
- The classical appearance in TOF is of a ‘boot shaped heart’ due to right ventricular hypertrophy and reduced central pulmonary markings (see Fig 4). The lung fields may be oligaemic.
ECG
- Signs of right ventricular hypertrophy and right axis deviation.
Echocardiography
- This is usually the only cardiac imaging required before surgery. The intracardiac abnormalities, degree and site of RVOTO and the coronary anatomy can be demonstrated.
- 25% of children with TOF have a right-sided aortic arch that may be been on CXR and confirmed by ECHO – this is important as palliative pulmonary shunts are easier to place on the opposite side to the aortic arch.
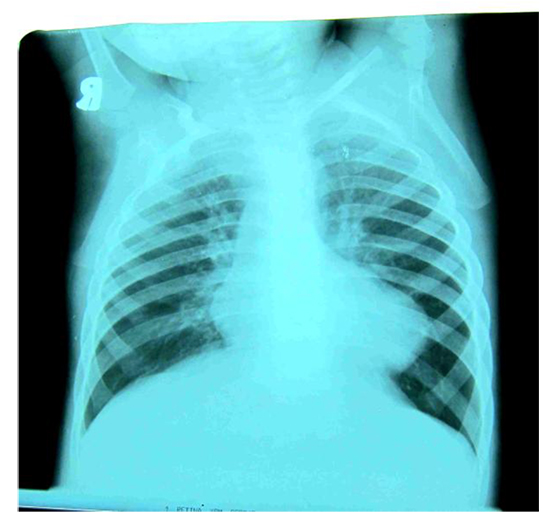
Figure 4. CXR showing ‘boot shaped’ heart in a child with TOF
MEDICAL MANAGEMENT
Before definitive corrective surgery is carried out, the child may be managed medically.
In the child with hypercyanotic spells, propranolol may be prescribed, usually in a dose of 0.5mg.kg-1 tds increasing to 1-1.5mg.kg-1 tds until symptoms are controlled. This reduces the spasm of the sub pulmonary infundibulum.
If the pulmonary blood flow is severely limited or increasing doses of propranolol fail to control spells, a palliative shunt may be required or corrective surgery expedited. Rarely, a child with severe spells may require intubation and ventilation, intravenous esmolol and phenylephrine prior to surgery (see below).
PALLIATIVE SURGERY – MODIFIED BLALOCK TAUSSIG SHUNT
A palliative shunt used to be the treatment of choice but is now rarely performed as the results of early corrective surgery have improved. However, certain patients may benefit from a palliative shunt in the first weeks or months of life:
- Newborns with duct dependent pulmonary circulation (pulmonary atresia)
- Marked RVOTO and cyanosis
- Very small pulmonary arteries
- Anomalous coronary artery anatomy
The shunt aims to achieve a postoperative target saturation of approximately 80-85% in air to allow the child to grow adequately before definitive surgery is undertaken. Signs that the child is out-growing the shunt are increasing cyanosis, and a return to earlier symptoms of breathlessness, fatigue and failure to thrive.
The modified Blalock Taussig (mBT) shunt is formed by placing a 3-4 mm Gortex tube graft end-to- side with the subclavian artery and the right or left pulmonary artery, depending on the position of the aortic arch and the anatomy of the pulmonary arteries (see Fig 5). The pulmonary arteries may be continuous with each other, or discontinuous (termed confluent/non-confluent), or one may be larger than the other. Flow provided by a palliative mBT shunt may encourage small pulmonary arteries to grow. Surgery may be performed via a thoracotomy or sternotomy depending on the surgical requirements.
Some centres perform transcatheter interventions such as ballooning or stenting of the RVOT in place of a mBT shunt prior to definitive surgery, but this is not common practice.
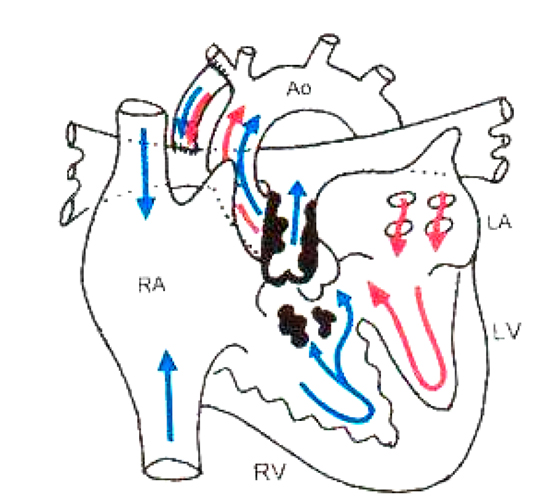
Figure 5. Modified Blalock Taussig shunt in TOF Reproduced with kind permission from: Pediatric Heart Surgery (A Ready Reference for Professionals). L. Eliot May www.maxishare.com
Anaesthesia for mBT shunt
Anaesthesia for a mBT shunt should proceed carefully in a child with uncorrected TOF, and preparations should be made to manage a hypercyanotic spell during surgery (see below). If the child has duct dependent pulmonary circulation, ductal patency should be maintained with prostaglandin infusion intraoperatively. The shunt is usually performed via a thoracotomy, but occasionally shunt insertion may be performed on bypass if the anatomy is difficult or pulmonary blood flow is critical.
The requirements for anaesthesia include:
- A unit of blood. This should be cross-matched and available, but is not usually required.
- Balanced anaesthesia, with intubation and ventilation and postoperative intensive care. Our practice is to provide intraoperative analgesia with fentanyl up to 10mcg.kg-1 and a postoperative morphine infusion.
- Full invasive monitoring with central and arterial access. The surgeon will clamp the subclavian artery during the shunt insertion, so the radial artery on that side should be avoided – femoral arterial access is preferable.
- For non-bypass cases, a small dose of heparin (100iu.kg-1) is required before the side-biting clamp is placed on the pulmonary artery.
- Inotropic support with an adrenaline infusion is occasionally required to maintain cardiac output after the shunt has been opened. Increased infundibular spasm is not a concern in this situation and the pulmonary infundibulum has been bypassed by the shunt (see hypercyanotic spells below)
The inspired oxygen should be reduced to 30% before the shunt is opened and an arterial blood gas taken to assess the adequacy of the shunt.
- The ideal situation – SpO2 80-85% with systolic blood pressure >80mmHg.
- Shunt too small or kinked – SpO2 <70% despite an adequate blood pressure
- Shunt too large – SpO2 100%, baby acidotic with low SBP despite inotropes. In this situation there is excessive pulmonary blood flow and inadequate systemic perfusion. The surgeon may partially occlude the shunt with a clip, or may need to replace it with a smaller shunt.
Postoperative care of a baby with a mBT shunt
Postoperatively, the continuous shunt murmur may be heard in the subclavian area, and the shunt flow should be confirmed using ECHO. A low-does heparin infusion is started, provided there is no postoperative bleeding, followed by aspirin after a few days.
Anaesthesia for a child with mBT shunt
A child with a mBT shunt may present for further cardiac or non-cardiac surgery. The mBT shunt is prone to obstruction, a complication that may be life threatening. The pulmonary blood flow is dependent on a patent shunt and adequate systemic perfusion.
- Check shunt patency preoperatively – SpO2 >80%, shunt murmur, ECHO if required.
- Avoid preoperative dehydration, consider intravenous fluids preoperatively.
- Maintain blood pressure during surgery. Consider a fluid bolus if the saturation falls.
- Careful postoperative monitoring.
CORRECTIVE SURGERY
Complete repair of TOF consists of closure of the VSD and relief of the RVOTO via median sternotomy on cardiopulmonary bypass. It is ideally performed in the first year of life, usually at 4-6 months of age. Some centres undertake corrective surgery in the first few weeks of life, but the outcome benefits are unclear at present. Good outcomes may still be obtained after repair of TOF in adolescence or adulthood, but there is increased risk of perioperative morbidity and mortality, mainly due to increased bleeding and ventricular dysfunction.
A trans-atrial approach is taken to repair the VSD using a bovine pericardial patch. Relief of the RVOTO depends on the level of obstruction. Ideally, integrity of the pulmonary annulus should be preserved if possible. Approaches to reduce the RVOTO include:
- Resection of muscle bundles in the pulmonary infundibulum via a transatrial approach or through an incision in the pulmonary artery
- RVOT patch, leaving the pulmonary annulus intact
- Transannular patch across the pulmonary outflow tract
- Pulmonary valvotomy or valvectomy
- Patch enlargement of the distal pulmonary arteries
A pulmonary conduit or homograft from the right ventricle to pulmonary artery may be required in children with pulmonary atresia or coronary abnormalities.
Adequacy of the repair is assessed by intraoperative transoesophageal echocardiography (to check for residual VSD and degree of residual RVOTO), or direct pressure measurements (ideally the right ventricular pressure should be less than 2/3 systemic).
Anaesthesia for repair of TOF
Specific considerations are those relating to the management of cardiopulmonary bypass in infants, the management of hypercyanotic spells, separation from bypass and postoperative intensive care management of patients with possible right ventricular dysfunction and junctional ectopic tachycardia.
Cardiopulmonary bypass in infants with TOF
The general approach to an infant undergoing cardiopulmonary bypass in our institution includes:
- Induction of anaesthesia with sevoflurane, maintenance anaesthesia with isoflurane, including during bypass
- Muscle relaxation with bolus dose atracurium.
- Intraoperative analgesia – fentanyl (total during the case 30-50 mcg.kg-1)
- Invasive monitoring, including central, arterial and a diagnostic monitoring line
- Transoesophageal echocardiography
- Routine anticoagulation with heparin 300-400 iu.kg-1
- Crossmatched blood (2-3 units), and blood products (platelets 10 m.kg-1 and cryoprecipitate 5-10 ml.kg-1)
- Antifibrinlolytic drugs (tranexamic acid 10-30 mg.kg-1)
- Inotropic support – milrinone 50 mcg.kg-1 loading dose, 0.375-0.5 mcg.kg-1.min-1; low dose adrenaline or noradrenaline as required (0.02-0.1 mcg.kg-1.min-1)
- Cooling to 32-34°C during bypass, maintenance haematocrit during bypass >21, 30 before separation from bypass
- Modified ultrafiltration post bypass to reduce total body water and increase haematocrit to aid blood conservation
Management of a hypercyanotic spell during anaesthesia
A history suggestive of hypercyanotic spells and use of propranolol should be sought in any child with TOF presenting for surgery. These patients may benefit from an oral sedative premed such as midazolam 0.5 mg.kg-1 30 minutes preoperatively or chloral hydrate 50 mg.kg-1 one hour preoperatively. In the very brittle patient, drugs to treat a hypercyanotic spell should be prepared in advance, heparin should be drawn up, and the surgical team should be standing by.
A hypercyanotic spell presents with rapidly falling saturation in response to surgical or other stimulation. ECG changes suggestive of ischaemia may occur and the blood pressure may fall. The precise pathophysiology of a hypercyanotic spell is unknown, but is thought to be due to muscular spasm in the pulmonary infundibulum leading to worsening RVOTO and increased right to left shunt across the VSD. Adrenaline and dopamine are β1 agonists and increase infundibular spasm. They worsen cyanosis by increasing shunt across the VSD and should be avoided in the perioperative period in children with TOF. Phenylephrine and noradrenaline are pure agonists. They increase SVR and reduce right to left shunt and should be used to treat cyanotic spells.
Management of a hypercyanotic spell should include general measures to reduce infundibular spasm, improve oxygenation and increase cardiac output, and specific measures to reduce right to left shunt and/or infundibular spasm:
- Increase inspired oxygen, check position of the tracheal tube
- Fluid bolus 10-20 ml.kg-1
- Deepen anaesthesia, give fentanyl bolus 1 mcg.kg-1
- Phenylephrine 1 mcg.kg-1 bolus up to 5-10mcg.kg-1. If repeated boluses required and central access available, start noradrenaline infusion 0.1 mcg.kg.min-1.
- Esmolol infusion may be considered to reduce infundibular spasm (starting dose 25mcg.kg-1.min-1)
Separation from bypass and routine postoperative intensive care
The child will often require inotropic support following corrective surgery for TOF. An infusion of a phosphodiesterase inhibitor such as milrinone is ideal, with or without a low dose of noradrenaline to maintain blood pressure. The right ventricle should be kept well filled to minimise residual RVOTO. Electrolyte imbalance should be avoided and serum potassium maintained close to 5 mmol.l-1 to avoid cardiac arrhythmias. Normothermia should be maintained (avoid hyperthermia). Bleeding should be controlled surgically, but blood products may also be required. Thromboelastography is useful to guide the use of blood products. Heart block may occur as a complication of surgery but is uncommon.
Postoperative ventilation is required until the child is warm, well perfused, and there is minimal bleeding (few hours to a few days, see below). Most children have an uncomplicated course and are discharged home within a week of surgery.
Postoperative restrictive physiology and junctional ectopic tachycardia (JET)
Following the repair of TOF, the right ventricle may exhibit signs of ‘restrictive’ physiology. This is due to systolic and diastolic myocardial dysfunction, associated with difficulty in achieving myocardial protection of the hypertrophied right ventricle. The right ventricle becomes ‘stiff’ and is unable to relax to accept the atrial volume during atrial systole. This is seen on Doppler echo as antegrade pulmonary artery flow in diastole. Restrictive physiology is associated with low cardiac output, renal impairment and pleural effusions. It is difficult to predict which patients will develop restrictive physiology, but this is more common in children with severe RVOTO who require insertion of a transannular patch. The patient should be managed with adequate filling, phosphodiesterase inhibitors and inotropes as described above. Diuretics and peritoneal dialysis may be required.
Junctional ectopic tachycardia (JET) may be seen after any cardiac operation, but most commonly after surgery close to the bundle of His, as in TOF repair. JET may be difficult to treat, and will exacerbate the effects of right ventricular dysfunction and low cardiac output.
JET should be treated aggressively, including:
- Cooling to 34°C
- Sedation and muscle relaxation to reduce the work of breathing
- Magnesium sulphate 25-50 mg.kg-1 loading dose, repeated as necessary to maintain plasma magnesium in the high normal range (1.5-2.5 mmol.l-1)
- Amiodarone infusion if required (25 mcg.kg-1.min-1 for 4 hours, then 5-15 mcg.kg-1min-1 (max1.2g/24 hour)
Postoperative restrictive physiology with or without JET usually resolves within 2-3 days, but ventricular dysfunction may recur and be a long-term problem. Restrictive physiology is associated with increased duration of postoperative ventilation, intensive care stay and in-hospital morbidity.
LONG TERM PROGNOSIS FOLLOWING TOF REPAIR
Prognosis has improved dramatically in recent years with improved anaesthesia, surgical and cardiopulmonary bypass techniques and postoperative intensive care. Early survival of 95-99% is expected. Long-term prognosis depends on the presence of pulmonary incompetence. Current surgical practice aims to preserve the pulmonary valve if possible, and mild residual RVOTO is tolerated.
The long-term effects of pulmonary incompetence include progressive exercise intolerance, right heart failure, arrhythmias and sudden death, often seen 10-20 years after complete repair. Prognosis in these patients is greatly improved by insertion of a pulmonary homograft or valved conduit, or in selected cases, by percutaneous valve implant in the angiography suite. Pulmonary valve implantation reverses ventricular enlargement and reduces arrhythmias. All children are closely followed up in adolescence; those with minimal residual defect may have completely normal activity, those with pulmonary incompetence and right ventricular enlargement have restricted exercise tolerance. Timing ofsecondary surgery for these patients remains controversial, but should take place before they develop severe complications associated with right ventricular dilatation.
SUMMARY
- TOF is the most common form of cyanotic heart disease
- The clinical presentation depends on the severity of RVOTO
- A ‘tet spell’ during surgery is best treated with volume loading and alpha agonists
- Long-term outcomes after corrective surgery are generally good; some patients require secondary surgery for pulmonary incompetence and right ventricular dilatation.
ANSWERS TO QUESTIONS
- The anatomical features of Tetralogy of Fallot may include
- Ventricular septal defect – True
- Coarctation of the aorta – False
- Aberrant pulmonary venous drainage – False
- Abnormal coronary artery anatomy – True
- Right ventricular hypertrophy – True
- Treatment prior to full correction may include
- Pulmonary artery banding – False
- Beta blocking drugs – True
- A prostaglandin infusion – True
- Diuretics – True, Diuretics may be required for a ‘pink tet’ where there is minimal RVOTO and the child mainly shunts left to right across the VSD to develop pulmonary congestion.
- Features of a hypercyanotic spell are
- Irritability – True
- Back arching – False
- 2:1 heart block – False
- Cyanosis – True
- Death – True
- Management of a Tetralogy ‘Spell’ may include
- Morphine – True
- Limiting inspired oxygen – False
- Increasing inspired oxygen – True
- Sodium nitroprusside – False
- Phenylephrine – True
REFERENCES and FURTHER READING
- Aptiz C, Webb G, Redington A. Tetralogy of Fallot. Lancet 2009 374: 1462-71
- Singh V. Tetralogy of Fallot: surgical perspective, treatment and management. E-Medicine http://emedicine.medscape.com/article/904652 (accessed 8th April 2011)
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/