Basic Sciences

The aim of this tutorial is to describe the physiology of the normal red blood cell and two of the most common abnormalities. Before reading this tutorial, consider:
- What is the structure of red blood cells?
- How does haemoglobin carry oxygen?
- What is different about fetal haemoglobin?
- What is the cause of sickle cell disease and thalassaemia?
The red blood cells
The red blood cells or erythrocytes are formed in the bone marrow by the process of erythropoeisis. Stem cells, under the influence of erythropoietin, develop into erythroblasts, at which point they start to synthesise haemoglobin. Mature erythrocytes are biconcave discs measuring 7.5m (microns or thousandths of a millimetre) in diameter. Their thick outer rim measures 2.5µm and their thinner centre only 1m. The shape of the disc confers three important properties:
- Deformability, allowing it to squeeze through narrow capillaries with out disrupting its cell membrane
- Enhances diffusion of gases into out of the cell by virtue of a greater surface area and shorter distance to the central regions than a sphere of the same volume
- Reduces wall tension as the cells swell after taking up carbon dioxide from the tissues.
Each red cell has a volume of 80-100fl (femtolitres, 10-15 l) and contains approximately 30pg (picograms, 10-12 g) of haemoglobin. In healthy individuals, there are normally around 5 x 1012 red cells per litre of blood and each litre contains 130-160g haemoglobin in males and 110-140g in females. Erythrocytes survive in the circulation for approximately 120 days and as a result, approximately 1% are replaced each day. This equates to a cell turnover of between 2-3million per second! As the erythrocytes age they become smaller and stiffer and are removed from the circulation by macrophages in the spleen, liver and bone marrow. Virtually all the components resulting from breakdown of the cells are recycled.
Red blood cells contain the enzyme carbonic anhydrase which catalyses the reaction between carbon dioxide and water to bicarbonate, thus enabling the transport of large quantities of carbon dioxide from the tissues to the lungs.
Haemoglobin and the carriage of oxygen
Haemoglobin is the constituent of the red blood cell that combines with oxygen and consists of two parts, haem and globin. Haem is a complex of a porphyrin ring and iron in the ferrous (Fe2+) state. Iron is an essential part of haemoglobin and when deficiency occurs due to dietary inadequacy or loss through chronic haemorrhage, anaemia results. Haem is conjugated with globin, a polypeptide, which consists of four subunits. Adult haemoglobin (HbA) comprises two alpha () and two beta () polypeptides. Differences in the polypeptide chains account for a number of haemoglobin variants, which alter the body’s capacity for transporting oxygen. These will be discussed later in the tutorial.
The amount of oxygen the blood carries is described as the oxygen content of blood. Although the vast majority of oxygen is carried bound to haemoglobin, a very small amount is dissolved in the plasma. The total amount of oxygen carried can be calculated by adding these two amounts. Under normal circumstances, as blood leaves the lungs, haemoglobin is almost fully saturated with oxygen and each gram of haemoglobin contains 1.39ml of oxygen. However, by the time it reaches the systemic circulation this has fallen slightly due to the addition of a small volume of venous blood from the pulmonary and coronary circulations. Therefore in the arterial circulation, one gram of haemoglobin is around 98% saturated and contains 1.34ml of oxygen. The amount of oxygen dissolved in the blood is proportional to the partial pressure. At 37 degrees Celcius, 0.23ml oxygen dissolves in each litre of blood per kPa. Therefore the oxygen content of blood can be calculated from the equation below and highlights the multiple factors required to optimise the ability of the blood to carry oxygen.
- O2 content of blood (l) = (Hb x 1.34 x SaO2) + (0.23 x PaO2)
- = (150 x 1.34 x 98%) + (0.23 x 13)
- ≈ 20.3 ml/l
Oxygen combines reversibly with the ferrous ion in the haemoglobin molecule to form oxyhaemoglobin.
- Hb + O2 ↔ HbO2
In the lungs, where the partial pressure of oxygen is high, the forward reaction oxygenating haemoglobin is favoured. In the tissues where the partial pressure of oxygen is low the reverse reaction reducing haemoglobin is favoured. Thus haemoglobin combines with oxygen in the lungs and releases it at the level of the tissues. Both the oxygenation and reduction of haemoglobin are extremely rapid reactions taking less than 0.01 seconds. The relationship between the saturation of haemoglobin and the partial pressure of oxygen in the blood is described by the oxygen dissociation curve.
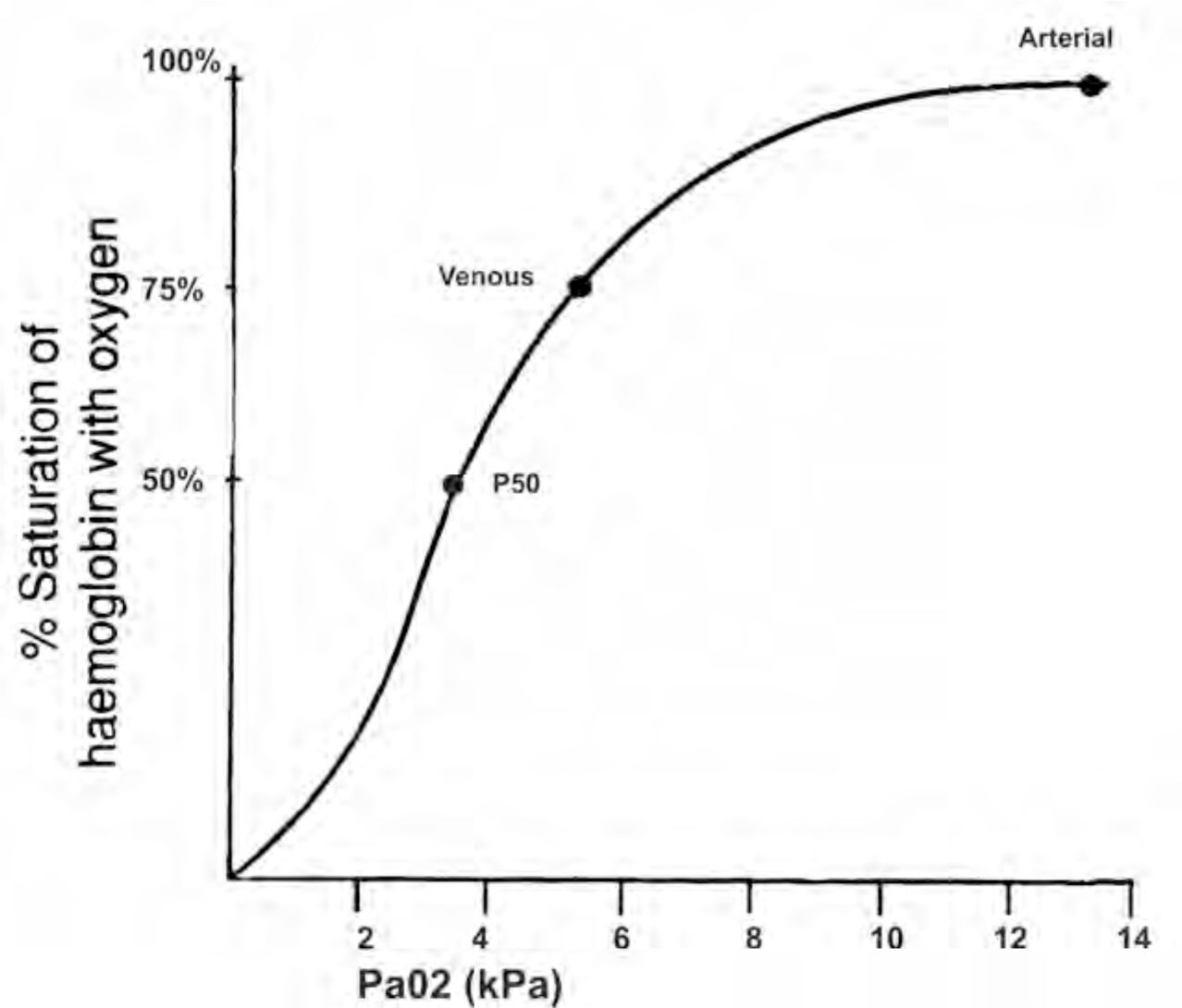
The curve demonstrates that at a normal PaO2 (13kPa), haemoglobin is approaching 100% saturation. The curve has a sigmoid shape owing to the cooperative binding of haemoglobin with oxygen. When one molecule of oxygen binds to the ferrous iron, steric reactions cause movement of the globin polypeptide chains. This facilitates the binding of further oxygen molecules. Thus at low oxygen tensions it is initially difficult for haemoglobin to combine with oxygen but this process gets much easier as more oxygen is available and as more oxygen combines with haemoglobin i.e. at the higher oxygen tensions available in the lungs. This is evident by the steep rise in the curve, until it plateaus as it approaches 100% saturation. The steep section of the curve is extremely important as it demonstrates that below 92% saturation the drop in saturated haemoglobin falls very quickly per unit partial pressure of oxygen.
A number of important points are shown on the curve. At 13kPa, the partial pressure of oxygen in the arteries, haemoglobin is nearly 100% saturated. At 5.3kPa, the partial pressure of oxygen in venous blood, haemoglobin is only 75% saturated. The third point demonstrated on the curve is the P50 value. This equates to the partial pressure of oxygen in the blood at which 50% of the haemoglobin is saturated. Under normal conditions this is 3.6kPa. It is useful when looking at how certain conditions affect the oxygen dissociation curve.
The dissociation curve can be shifted to the right or left by a number of factors. The curve is displaced to the right by;
- Increased hydrogen ions (reduced pH)
- Increased carbon dioxide
- Increased temperature
- Increased DPG
A right shift in the curve means that haemoglobin releases its oxygen relatively easily. This occurs during an acidosis. Deoxyhaemoglobin binds more actively with hydrogen ions than oxyhaemoglobin, thus as the hydrogen ions rise and the pH consequently falls, haemoglobin’s affinity for oxygen also falls. The reverse also occurs. There will also be a rise in the P50 value, as only 50% of the haemoglobin molecules are saturated at a higher partial pressure compared to normal.
Carbon dioxide has a similar effect on the position of the dissociation curve, increases shifting it to the right and decreases to the left. This comes about as a result of an increase in PCO2 causing an increase in hydrogen ions (decrease in pH) and also a decreased affinity of haem for oxygen. In the lungs, the converse is true. The influence of hydrogen ions and CO2 on haemoglobin affinity for oxygen is known as the Bohr effect.
2,3 diphosphoglycerate (DPG) is formed in red blood cells as a product of glycolysis. It is a highly charged ion and when it combines with the beta chain of haemoglobin, it displaces oxygen and again shifts the curve to the right. The conditions that cause a rightward shift in the curve thus encouraging the unloading of oxygen at relatively low partial pressures of oxygen can be summarised by thinking about the exercising muscle. During exercise there is an increase in temperature, hydrogen ions, CO2 and DPG in the tissues, all the conditions associated with a rightward shift in the curve to help deliver oxygen to the active muscles.
Fetal haemoglobin
Haemoglobin in the fetus (HbF) is structurally different from the adult form (HbA). As described earlier haemoglobin A has two alpha and two beta globin chains. However in haemoglobin F the two beta chains are replaced by two gamma chains. This structural difference means that fetal haemoglobin binds less avidly with DPG and, as we know, DPG reduces the amount of oxygen that combines with haemoglobin. Consequently fetal haemoglobin has a higher affinity for oxygen than adult haemoglobin. This is demonstrated by a leftward shift in the oxygen dissociation curve (and the lowering of the P50). This difference in haemoglobin physiology enables the fetus to extract oxygen from maternal blood at the placenta. Adult haemoglobin quickly replaces fetal haemoglobin after birth thus correcting the position of the curve and allowing the baby to utilise oxygen normally. 2.5% of adult circulating haemoglobin is HbF.
Haemoglobin variants
There are a range of genetic variants which lead to abnormal haemoglobin production. This tutorial will look at the most common variants:
- Sickle cell disease and sickle cell trait
- Thalassemia
Sickle cell disease
This is a haemoglobinopathy with autosomal recessive inheritance. The genetic defect leads to the substitution of valine for glutamine at position six on globin’s beta chain, creating HbS rather than HbA. The gene is carried by 10% of people of African extraction, however it is also seen in Indian, Arabian, Italian and Greek populations.
There are a number of genotypes that make up the range of patients suffering from sickle cell disease. The two most common are:
- HbSS – patients who are homozygous and have sickle cell disease
- HbSA- patients who are heterozygous and have sickle cell trait
At low partial pressures of oxygen (5-5.5kPa) deoxygenated HbS becomes insoluble. It forms long crystals called tactoids which cause red cells to become sickle shaped and rigid. The distorted cell shapes and associated increased blood viscosity promote venous stasis. This leads to local blood vessel obstruction causing ischaemia and tissue infarction. These events are termed a sickle cell crisis. Other factors that increase the rate of sickling are acidosis, sepsis, hypothermia and cellular dehydration. The process of red cell sickling is reversible until potassium and water are lost from the cell. Then the cell membrane becomes brittle and the cell sickles permanently. These cells have a decreased survival time, of only 10-20 days. Therefore patients with sickle cell disease suffer from a chronic anaemia (haemolytic) and are often jaundiced (due to conversion of haem to bilirubin).
A sickledex test can confirm the presence of haemoglobin S and electrophoresis can differentiate between trait and disease. Electrophoresis can also determine the proportion of HbA to HbS in the blood. Patients who have sickle cell trait are also at risk of a sickle cell crisis but only at very low partial pressures of oxygen as their erythrocytes only contain 20 to 45% HbS.
Clinical features
Sickle cell crises usually occur from six months of age as this is when adult haemoglobin replaces fetal haemoglobin. Some patients maintain a higher than normal proportion of HbF into adult life, which improves their clinical condition. Sickle cell disease affects a many of the body’s systems as outlined below.
Haematological
- Chronic haemolytic anaemia
- Splenic infarction
- Aplastic anaemia secondary to bone marrow infarction
- Increased susceptibility to infection
- Acute sequestration syndrome in the liver or spleen causing a dramatic acute fall in haemoglobin
Respiratory
- Acute chest syndrome characterised by pleuritic chest pain, dyspnoea and haemoptysis.
- Recurrent episodes can lead to pulmonary hypertension and chronic respiratory failure.
Renal
- Haematuria and renal failure
Musculoskeletal
- limb shortening and deformity, including the neck and mandible
Skin
- Ulceration
Genitourinary
- Priaprism
Neurological
- Acute brain syndrome, characterised by confusion, recurrent episodes can result in permanent damage
Gastrointestinal
- Liver sequestration syndrome
- Haemosiderosis associated with repeated transfusions
- Gallstones secondary to chronic haemolysis.
Not surprisingly patients with sickle cell disease frequently require surgery and anaesthesia. As a result of their disease, they are often anaemic and have existing organ damage. In severely anaemic patients, exchange transfusion is preferable to simple transfusion, but it must be remembered that patients have usually received multiple blood transfusions creating abnormal antibodies, thereby extending the cross matching time. Preoperative tests can be used to assess the extent of organ damage and include urea and electrolytes, liver function tests and an ECG. Heavy premedication with opioids should be avoided, where possible, to prevent hypoventilation prior to theatre. Where opioids are essential for example due to abdominal pain, supplementary oxygen should be given. The anaesthetic aims are the avoidance of factors that would precipitate sickling and are the same as underpin every anaesthetic, namely the avoidance of hypoxia, hypercarbia, acidosis, hypothermia and low flow states. Tourniquets and vasoconstrictors should be avoided and care with positioning to prevent regional low flow and acidosis. Optimisation of fluid balance is essential and where possible invasive monitoring should be considered. Patients should be actively warmed, and antibiotics given especially if the patient is asplenic.
Regional anaesthesia should be considered as it has a number of advantages for patients with sickle cell disease. The sympathetic block will cause vasodilatation leading to increased peripheral perfusion, it offers excellent analgesia in the initial postoperative period and it avoids any difficulties surrounding intubation in patients with skeletal abnormalities. However these advantages need to weighted against the serious side effects of hypotension and hypoperfusion caused by regional blockade.
Thalasseamia
Thalassaemia is an inherited disease of haemoglobin production. Patients with the disorder have a reduced or absent production of globin chains. Many point mutations have been discovered which lead to defective thalassaemia genes. It is commonly found in people of Mediterranean origin.
Thalassaemia
This is where the production of the beta globin chain is impaired. The production can be impaired to varying degrees. Excess alpha chains combine with any other chain that is available and precipitate in the cells leading to ineffective erthyropoeisis and haemolysis.
Thalassaemia trait (minor)
This is the heterozygous form of the condition. Patients are usually asymptomatic, but may present with a mild hypochromic, microcytic anaemia. This can be differentiated from iron deficiency as the serum iron and ferritin levels are normal. Electrophoresis would reveal an increased level of Hb A2 (two alpha chains) and HbF.
Thalassaemia intermedia
As the name suggest the clinical features of this disorder lie somewhere between thalassaemia minor and major. Patients are usually symptomatic from a moderate anaemia, but do not require regular transfusions. It is believed to be caused by a combination of mild and thalassaemias resulting in less alpha chain precipitation thus less haemolysis.
Thalassaemia major (Cooley’s anaemia)
This is the homozygous state, leading to markedly reduced or absent globin chain production. Children present with failure to thrive, anaemia, jaundice or hepatosplenomegaly and bone expansion secondary to extramedullary erythropoesis. Patients suffer from a severe anaemia and require regular blood transfusions, which commonly leads to complications due to iron overload. Transfusion haemosiderosis causes damage to the endocrine glands, liver, pancreas and myocardium.
Thalassaemia
There are four genes encoding for alpha chain production, located on chromosome 16. In the absence of all four genes no alpha chains will be produced and this is incompatible with life. This condition is known as Hb Barts, babies are born stillborn with hydrops fetalis. Aside from Hb Barts thalassaemias are usually much better tolerated than thalassaemias. Three alpha gene deletions leads to a moderate anaemia, but patients are not usually transfusion dependent. However patients with only one or two gene deletions have mild anaemias and are usually asymptomatic.
Thalassaemia and anaesthesia
The minor thalassaemias do not pose too many problems during anaesthesia. Patients are usually anaemic, thus any other contributing causes to this require identification. thalassaemia major requires special consideration. A thorough pre-operative assessment is needed to establish the extent of any end organ damage caused by iron overload. It is common for patients with severe anaemia to suffer from a high output congestive cardiac failure, thus patients should be transfused preoperatively. As with with sickle cell disease, patients who have had repeated transfusions are likely to have developed antibodies that will prolong the time taken to cross match blood. Finally airway management and laryngoscopy may be made difficult as a result of the hyperplastic bone marrow causing overgrowth of the facial bones, in particular prominent maxillae, a sunken nose and thickened skull.
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/