Paediatric Anaesthesia

KEY POINTS
- Tracheo-oesophageal fistula (TOF) with or without oesophageal atresia (OA) has a worldwide incidence of 1 in 3000-4000 births, and most cases are diagnosed postnatally. Up to 50% of infants have other congenital anomalies in association with TOF/OA.
- Prior to surgical repair, a thorough preoperative assessment must be performed, paying close attention to the neonate’s respiratory and cardiac status including an echocardiogram to detect congenital heart disease lesions.
- Intraoperative considerations for TOF/OA repair include anticipation of difficulties with airway management, surgical technique, potential for blood loss, and plans for post-operative management.
- While outcomes in neonates with TOF/OA have improved significantly due to advances in surgical, anaesthetic, and critical care management, mortality rates remain high in low resource setting.
INTRODUCTION
Tracheo-oesophageal fistula (TOF) with or without oesophageal atresia (OA) is a congenital anomaly with an incidence of 1 in 3000-4000 births worldwide.1 While TOF may occur in isolation, up to 50% of infants have TOF in association with other congenital anomalies, the most common being congenital heart disease. In addition, up to 25% of TOF infants are diagnosed with VACTERL (Vertebral anomalies, imperforate Anus, Cardiac defects, Tracheo-oesophageal fistula, Renal agenesis, and Limb abnormalities, most often radial dysplasia).2 (See Figure 1)
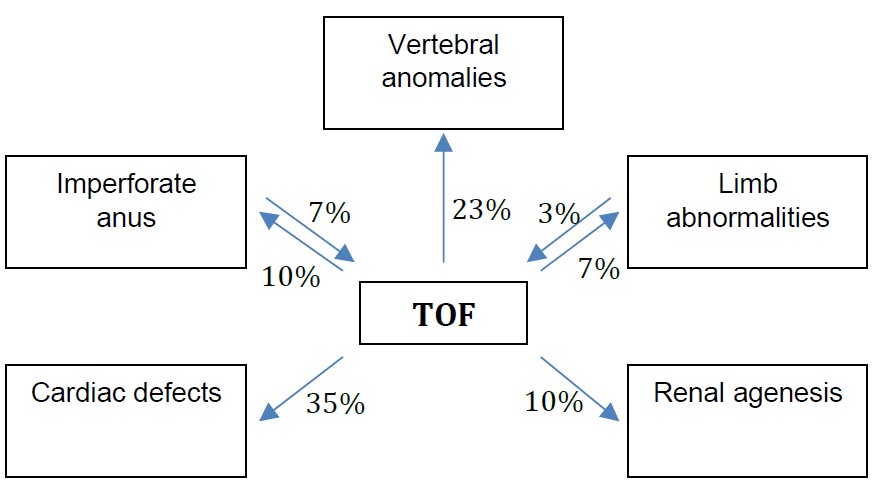
Figure1. Frequency of associated VACTERL syndrome defects and TOF2
Clinicians must recognize that children with syndromic TOF, especially those with coexisting complex congenital heart disease, will need additional surgeries and will be more complex to manage in the perioperative setting. Our ability as anesthesiologists to anticipate the challenges in managing infants preoperatively, intraoperatively and postoperatively plays an important role in their treatment and survival.
Anatomy of Tracheo-Oesophageal Fistula
During the fourth week of embryonic life, the trachea is differentiated from the oesophagus. If there is an incomplete separation of the trachea from the floor of the foregut, tracheo-oesophageal malformations are formed. According to the Gross classification system, there are six anatomic variations of TOF with and without OA (Types A-F, Figure 2).3 In type C, the most common lesion which occurs in more than 90% of cases, a fistula exists between the trachea and the lower oesophageal segment above the carina while the upper oesophageal pouch ends blindly in the mediastinum.3
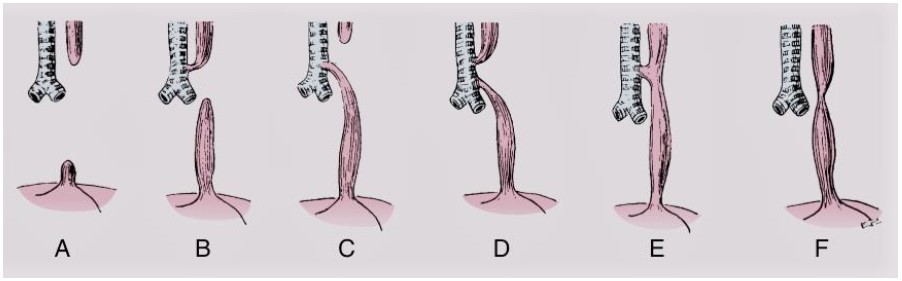
Figure 2. Variants of tracheo-oesopageal fistula with and withouth oesophageal atresia3. Reproduced with permission from Elsevier
Diagnosis
Unlike many other congenital anomalies, TOF is not easily diagnosed prenatally. The most common prenatal ultrasound finding associated with TOF/OA is polyhydramnios secondary to oesophageal obstruction and the inability to swallow amniotic fluid. However, polyhydramnios is a nonspecific sign and is caused by congenital anomalies less than 20% of the time.3 As a result, most TOF/OA cases, in both low and high resource settings, are diagnosed postnatally when patients present with excessive salivation and choking with feeds. To confirm the presence of OA, a suction catheter is inserted orally or nasally into the neonate and is unable to be passed more than 10cm into the esophagus. Diagnosis can then be confirmed by chest radiograph which demonstrates the catheter in the blind upper pouch on chest radiograph (Figure 3).
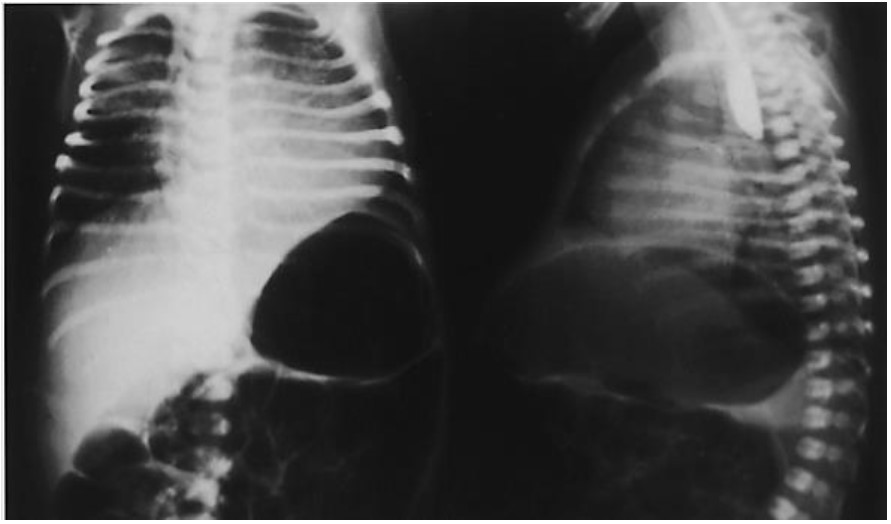
Figure 3. Type C lesion with gastric distention. Blindly ending esophagus on lateral view. Reproduced with permission from Elsevier 3
In the low-resource settings patients are referred from the periphery to tertiary health care facilities for definitive treatment which can take several days. This time period is frequently associated with aspiration, pneumonia and sometimes sepsis.4 Depending on the physical condition of the neonate on arrival at the tertiary health care facility, it is not uncommon for operative repair to be delayed from 3 days to 2 weeks after birth. Ideally, during this time period, a nasogastric tube passed into upper pouch is suctioned intermittently using low pressure suction. Fluid and electrolyte imbalances are corrected and sepsis is managed with antibiotics. Prior to definitive repair especially for more complex anatomies, it is sometimes necessary to perform a cervical oesophagostomy for drainage of the upper pouch and place a gastrostomy tube for enteral feeding.4 The definitive repair is then performed at a later date after the neonate’s condition has stabilized.
PREOPERATIVE ASSESSMENT
It is not uncommon for neonates with TOF to present with respiratory complications as they are prone to aspiration. A chest radiograph may show infiltrates. Prevention and or treatment of pulmonary complications is imperative for reducing morbidity and mortality. To minimize the risk of developing aspiration pneumonitis, the following interventions are undertaken: all oral feeds are stopped, the neonate is kept in upright positioning, and intermittent suctioning of the upper oesophageal pouch is performed to decrease the accumulation of saliva.
A thorough preoperative assessment should focus on determining the presence and possible anesthetic implication of any coexisting congenital abnormalities, especially cardiac defects. The high prevalence of congenital heart disease makes a preoperative echocardiogram a necessity. Cardiac defects such as ventricular septal defect, atrial septal defect, tetralogy of Fallot are commonly associated with TOF and will impact the anaesthetic management. In addition, an echocardiogram can reveal aortic arch anomalies, which can affect surgical technique. As many as 5% of patients have been reported to have right-sided aortic arch requiring a left thoracotomy.5
While respiratory and cardiac preoperative assessments are essential, if a neonate has any VACTERL anomalies, diagnostic testing must be performed to assess of coexisting anomalies. If a sacral dimple is present it is beneficial to obtain a lumbar ultrasound to evaluate neuraxial anatomy especially if planning to place a caudal catheter for postoperative analgesia. Additionally, a renal ultrasound should be obtained to rule out hydronephrosis or other renal anomalies, which may also affect anaesthetic management.
INTRAOPERATIVE MANAGEMENT
Prior to surgical repair, a detailed intraoperative plan must be developed that takes into account surgical technique, airway management and potential for blood loss.
Surgical Technique
There are two main surgical approaches for TOF/OA repair: the traditional surgical approach via an open thoracotomy and the thoracoscopic approach. Thoracoscopy is minimally invasive and is performed with the use of small fiberoptic cameras by surgeons who have obtained specialized training in the technique. The primary advantage of thoracoscopy is reduction in musculoskeletal sequelae such as “winged” scapula. However, due to compression of the ipsilateral lung with operative pneumothorax to achieve an adequate working space, desaturation is common as is reduction in venous return as a result of direct compression of vascular structures such as the inferior vena cava and the right atrium. Carbon dioxide absorption results in hypercarbia and acidosis which may be poorly tolerated by these neonates.
For the open technique, which is the technique of choice in developing countries as equipment and training for thoracoscopic repair is not available, the patient is positioned in the lateral decubitus position. Usually, the infant is in left decubitus positioning for a right thoracotomy approach, unless there is evidence of a right-sided aortic arch in which case a left thoracotomy is performed. A total open repair can be accomplished as a one-stage procedure where the fistula is ligated and the oesophagus is primarily anastomosed. If the fistula is at the level of the carina, one lung ventilation may be necessary, predisposing the infant to hypoxemia, hypercarbia, and developing an increase in pulmonary vascular resistance. The lung should be carefully re-expanded to eliminate atelectasis before closure.
Anaesthestic Management
Anaesthetic management in the operating room should include routine monitors (EKG, blood pressure cuff, end tidal CO2, temperature probe, and one pulse oximeter unless a ductal-dependent cardiac lesion is present in which case two pulse oximeters are needed for the measurement of pre- and post-ductal saturations). If available, an arterial line for continuous hemodynamic monitoring and analysis of blood gases is helpful. Preferably the arterial line is placed in the left upper extremity, as the right arm is elevated with limited access during right thoracotomy. At least two peripheral IVs should be placed, and cross-matched blood should be available in the operating room. If there is a high likelihood of the need for parenteral nutrition postoperatively, central access must be obtained.
The goal during induction is to intubate the infant while minimizing distention of the stomach. If measures are not taken to avoid stomach distention, this can make it very difficult, if almost impossible to ventilate the infant and can lead to hemodynamic collapse. There are several ways to secure the airway. The conservative approach involves an awake intubation to avoid positive pressure mask ventilation, however, attention must be paid to the possibility of increased intracranial pressure and intraventricular hemorrhage in the premature infant. Another technique is to induce general anesthesia via the inhalation route while maintaining spontaneous ventilation and then have the surgeon perform a rigid bronchoscopy. The role of the bronchoscopy is to determine the exact location and size of the fistula and to assist with placement of the endotracheal tube (ETT) distally to the fistula but above the carina.2,3 If fiber-optic bronchoscopy (FOB) is available, after ETT placement, FOB can be used to properly position the ETT tube. The carina is identified and then the ETT is pulled back (with FOB inside ETT) until the fistula is visualized, then the ETT is advanced just distal to the fistula. Placement of the ETT can be challenging especially when the TOF is located at the level of the carina. In such cases a Fogarty catheter can be placed into the fistula and inflation of the catheter balloon helps prevent gastric aspiration and inadvertent ventilation through the fistula.
Once positioned, there are various ways to confirm placement of the endotracheal tube in good position just distal to the fistula:
- Auscultation of breath sounds
- Right (or left) mainstem intubation with gradual withdrawal until bilateral ventilation is established, ideally about 1 cm above the carina
- Chest radiograph
- Ultrasound
- If a gastrostomy is present, bubbling versus lack of bubbling when the tube is above or below the fistula communication
Currently the need for gastrostomies varies depending on resource setting. In developed countries gastrostomies are not routinely performed because gas from the trachea may bypass the lungs and exit through the stomach leading to loss of effective ventilation. In low resource settings, however, where bronchoscopy may not be available, following inhalational induction and spontaneous respiration, the surgeon routinely performs a gastrostomy. This is then followed by endotracheal intubation under deep inhalational technique or using muscle relaxant technique and gentle manual ventilation. The initial gastrostomy allows gas to be vented out and thus prevents gastric distension and minimizes the risk of aspiration.
Once the airway is secured and appropriate intravenous and arterial access is obtained, the patient is positioned in the lateral decubitus position and pressure points are carefully padded. Maintenance of adequate oxygenation can be a major intraoperative problem. Accumulations of blood or secretions in the endotracheal tube can lead to airway obstruction, requiring frequent tracheal suctioning. Surgical airway manipulation and collapse of the upper lung due to use of retractors can also lead to episodes of marked hypoxemia. Close communication with the surgical team is of paramount importance, and intubation equipment should be readily available in case of accidental extubation and the need for emergent re-intubation.
POSTOPERATIVE CONSIDERATIONS
After TOF surgical repair via a thoracotomy, full term neonates without co-morbidities can be extubated postoperatively. Infants with other congenital anomalies or infants who require more complex TOF repairs may need postoperative intubation for a few days in the neonatal intensive care unit. A nasopharyngeal catheter is used for suctioning and is carefully taped or sutured in place so as to prevent insertion past the oesophageal anastomotic site.
In low-resource settings, patients are managed in the intensive care unit which is often a general ICU as neonatal intensive care units are not readily available.6 Postoperative ventilation is often done with a versatile ICU ventilator adapted for neonates and not specifically with a neonatal ventilator. This poses multiple challenges in terms of ventilator setting and adequacy of ventilation. In particular, the challenge of adequacy of ventilation may be compounded by erratic availability of arterial blood gas measurement and nursing staff not trained in neonatal critical care.
Regional anesthetic techniques such as caudally placed epidurals have been used for pain management, but in most places postoperative analgesia is maintained with opioids and paracetamol. In some developing countries, intercostal nerve blocks are performed by the surgeon.
Postoperative complications
While outcomes in neonates with TOF/OA have improved significantly due to advances in surgical, anaesthetic and critical care management, postoperative complications are still common. Early postoperative complications primarily include oesophageal strictures and anastomotic leaks. Some leaks may require surgical treatment if not able to close spontaneously. Oesophageal strictures may require multiple balloon dilations, but most early strictures respond to 2-3 dilations. Following a TOF repair, it is recommended that patients have endoscopic follow-up for up to three years. If there are signs of oesophagitis, follow up is extended by three more years and biopsies are obtained to determine if Barrett’s esophagus, a precancerous lesion, is present.7
Persistent gastroesophageal reflux is probably the most common long term problem and occurs more frequently after a delayed primary repair. If gastroesophageal symptoms are severe, surgical intervention may be required via Nissen Fundoplication. In addition, many TOF/OA patients have oesophageal dysmotility, which may lead to the development of oesophageal obstruction and growth failure.
A rarer complication, but one that can define long term outcomes after TOF/OA repair is tracheomalacia. Tracheomalacia can cause collapse of the airway resulting in stridor, apnea or recurrent pneumonia. Tracheomalacia tends to improve after the first 3 to 5 years of life, but if severe may require treatment with tracheopexy or aortopexy 7,8 .
Overall, mortality rates are less than 10% in developed countries. The Spitz system predicts the prognosis of patients with TOF/OA based on their birth weights and the presence or absence of major congenital heart disease (Table 1). Low birth weight infants who have cardiac anomalies have the highest morbidity and mortality rates.3

Table 1. Spitz survival rates, 2006.
In low-resource settings, mortality rates remain high and vary from 40% to 80%.6 The high mortality rates have been attributed to late presentation which is often associated with aspiration and pneumonia. Other factors which contribute to mortality include minimal supportive care such as lack of neonatal intensive care and parenteral nutrition for early feeding. Early postoperative complications include sepsis and respiratory failure. Late postoperative complications in survivors are the same as those described in developed countries. As with other congenital anomalies, children with TOF/OA require long term supportive multidisciplinary care with follow up to minimize further complications.
REFERENCES AND FURTHER READING
- Goyal, A; Jones, M.O; Couriel, J.M; Losty, P.D. Oesophageal atresia and trachea-oesophageal fistula. Archives of Disease in Childhood-Fetal and Neonatal Edition. 2006; 91(5) F381-F384.
- Hung O, Murphy MF. Management of the difficult and failed airway. 2ndedn. New York: McGraw-Hill Medical,2011.
- Davis PJ, Cladis PF, Motoyama EK. Smith’s anesthesia for infants and children. 8thedn. Philadelphia: Mosby Publishing, 2011.
- Adebo OA. Oesophageal atresia and 5rachea-oesophageal fistula: review of a 10 year personal experience. West AfrJ Med 1990; 9(3): 164- 169.
- Harrison MR, Hanson BA, Mahour GH, Takahashi M, Weitzman JJ, The significane of right aortic arch in repair of esophageal atresia and tracheoesophageal fistula. J PediatrSurg 1977; 12:861-869.
- Ameh EA, Bickler SW, Lakhoo K, Nwomeh BC, Poenaru D. Oesophageal Atresia. Paediatric surgery: A comprehensive Text for Africa. Global Help Publication 2010; 306-309.
- Kovesi T, Rubin S. Long-term complications of congenital esophageal atresia and/or tracheoesophageal fistula. Chest 2004; 126:915-925.
- Schalamon J, Lindahl H, Saarikoski H et al. Endoscopic follow-up in esophageal atresia-for how long is it necessary? J PediatrSurg 2003; 38:702-704.
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/