Paediatric Anaesthesia

QUESTIONS
Before continuing, try to answer the following questions. The answers can be found at the end of the article, together with an explanation.
- Which of the following statements is correct?
- Coarctation of the aorta (CoA) only occurs in adults.
- Women are seven times more likely to have coarctation than men.
- Once the CoA is corrected, cardiovascular symptoms disappear.
- If left unrepaired mortality approaches 90% by 55yrs of age.
- Describe the clinical differences between early and delayed presentation.
- Which of the following may occur as postoperative complications?
- Paralysis
- Hypotension
- Haematemesis
- Chylothorax
- Hoarse voice
INTRODUCTION
Coarctation of the aorta is a narrowing of the aorta resulting from an abnormal junction of the aortic isthmus and the arterial duct. It can present in the neonatal period, early childhood or in adulthood. It accounts for 5% of all congenital cardiac defects and mortality is greater than 80% if unrepaired.
DEFINITION AND ANATOMY
Coarctation literally means “a drawing together” and is a narrowing of the aorta (see Figure 1). It usually occurs distal to the left subclavian artery but can occasionally occur proximally. Coarctation of the aorta (CoA) is often described by the relationship of the coarctation to the ductus arteriosus (ligamentum arteriosum in adults). CoA may be ‘pre-ductal’, ‘juxtaductal’ or ‘postductal’. This is an arbitrary classification as the area of coarctation may shift in position as the aortic arch grows and thus the classification does not represent a true difference of origin, rather the stage of evolution of the CoA.
CoA can consist of an in-folding of the wall of the aorta, called a WAIST type lesion, an intimal defect, also known as a SHELF or DIAPHRAGM lesion or can be due to extensive arch hypoplasia termed TUBULAR HYPOPLASIA of the aortic arch. The extreme end of the coarctation is AORTIC INTERRUPTION, which describes complete separation of the lumen of the aorta into two segments with a connecting fibrous strand.
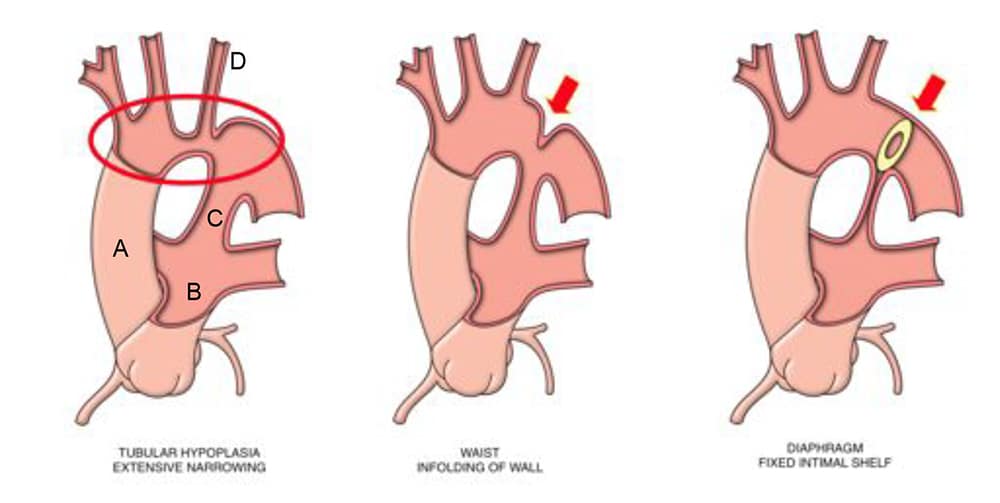
Figure 1: Different sites of narrowing in coarctation of the aorta

Gemma Price WCH ©
INCIDENCE AND ASSOCIATIONS
Coarctation occurs in 1 in 2000 live births in the USA and is the fifth most common congenital cardiac defect. It is more common in boys (M:F 1.7:1) and affects Caucasians seven times more than other races. 75% of children with CoA have another cardiac anomaly, most commonly patent ductus arteriosus (PDA), bicuspid aortic valve, ventricular septal defect (VSD) and mitral valve anomalies. CoA may also be associated with 22q11 deletion (thymic aplasia, cleft palate, hypocalcaemia, mild developmental delay), and hypoplastic left heart syndrome.
Other associations:
Turner’s syndrome. CoA is seen in up to 15% of patients. This is a chromosomal abnormality, 45XO, associated with short stature, webbed neck, widely spaced nipples, low hairline, small chin, prominent ears.
Kabuki syndrome. This is a genetic abnormality associated with developmental delay, joint laxity, cleft palate and characteristic facial appearance with arched eyebrows. It is associated with a hypoplastic isthmus and juxtaductal CoA in 25% of patients, also anomalous origin of one or both coronary arteries from the pulmonary artery.
Shone’s syndrome is a series of four abnormalities – supra-valvular mitral membrane, parachute mitral valve, subaortic stenosis and CoA. This is rare and associated with a poor prognosis due to a combination of both inflow and outflow obstruction to the left ventricle.
Abnormalities of the right heart are rarely associated with CoA.
AETIOLOGY
Two possible explanations exist regarding the aetiology of CoA; the haemodynamic theory and the abnormal duct theory. The first postulates that there is decreased blood flow into the aorta secondary to left heart obstruction in utero, which leads to under-development of the aorta at the isthmus. The second postulates that abnormal ectopic ductal tissue in the aortic wall causes constriction when ductal tissue constricts after birth. However, ductal tissue has not been observed at the site of all CoA.
Associated factors include:
- Genetic mutations – Caucasians are seven times more likely to have CoA.
- Environmental factors – There is a higher incidence of CoA in babies born in the autumn and winter months.
NATURAL HISTORY
Unrepaired, mortality from CoA approaches 90% by the age of 55yrs. The most common cause of death is cardiac failure (25%) followed by aortic rupture (21%), endocarditis (18%) and finally intracranial haemorrhage (12%). 10% of those with CoA have intracranial aneurysms within the Circle of Willis.
If repaired before the age of 14yrs the 20yr survival rate is 91%. If repaired after age 14yrs the 20yr survival rate is 79%.
During pregnancy there is a risk for aortic dissection or intracranial haemorrhage. Maternal mortality may be as high as 3-8%, even in those who have undergone repair. Thus all pregnancies should be treated as high risk. Continuing significant stenosis, whether native, residual or recurrent is a contraindication to pregnancy.
CLINICAL PRESENTATION
The presentation of CoA varies according to:
- Severity
- Presence of associated defects
- Extent of ductal patency
- Presence of collaterals
Early presentation
Children with severe CoA (usually pre-ductal CoA) present in the neonatal period with circulatory collapse, absent femoral pulses and signs of acute left ventricular failure (LVF). These neonates are said to have ‘duct-dependent systemic circulation’. Circulatory collapse coincides with closure of the ductus arteriosus and loss of lower body perfusion (which had been maintained by the pulmonary artery (see figure 1)).
When CoA is less severe, neonates may present with more subtle signs such as decreased femoral pulses noted at the post-natal check, hypertension, congestive cardiac failure, tachyopnoea, cyanosis, feeding difficulties and listlessness. Some neonates are diagnosed before birth by foetal echocardiogram, but the aorta is difficult to image in utero, so a confirmatory echocardiogram is required postnatally. CoA with associated defects may also lend to an earlier presentation, especially hypoplastic left heart syndrome.
Immediate management of a neonate presenting with severe CoA:
- Re-establish perfusion to lower body. Start prostaglandin E1 infusion to reopen the ductus arteriosus. (60 mcg/kg PGE1 in 50 ml 5% dextrose, IV infusion at 0.5-3 ml/hr i.e. 10-60 ng/kg/min).
- Treat LVF. Intubate, ventilate, start inotropic support (e.g. dopamine 3mg/kg in 50 ml 5% glucose, IV infusion at 5-10 ml/hr; i.e. 5-10 mcg/kg/min) and diuretics.
- Urgent surgery is required when these two strategies fail and/or there is still significant acidosis or anuria.
Children with moderate CoA may present in infancy or early childhood with failure to thrive, poor feeding, tachypnoea, lethargy and poor exercise tolerance due to LVF, or an incidental finding of upper limb hypertension. LVF should be treated before surgery is considered.
Late presentation
Patients with CoA may present in later childhood or adulthood with the incidental finding of upper limb hypertension or a murmur due to a bicuspid aortic valve. Blood pressure should always be taken in both arms as there may be an aberrant right subclavian artery which is distal to the coarctation. Patients may also present with symptoms of headache, fatigue or intermittent claudication. Occasionally, patients present more dramatically with untreated CoA, with aortic rupture, intracranial haemorrhage, left ventricular failure or features of endocarditis. CoA is this circumstance is usually post-ductal.
CLINICAL FINDINGS
Clinical findings vary according to severity and associated anomalies.
Early presentation – signs and symptoms:
Signs and symptoms of cardiogenic shock, left ventricular failure, and any associated cardiac and extra cardiac anomalies:
Observation:
- Tachyopneic
- Small for age
- Grey skin
- Lethargic
Palpation:
- Tachycardia
- Absent lower limb pulses
- Radio-radial and radio-femoral delay
- Prolonged capillary refill
Auscultation:
- Murmurs associated with associated VSD, aortic or mitral valve defects.
- Gallop rhythm
- Lung crepitations
- Upper limb hypertension. A gradient of 35mmHg between upper and lower limb BP is significant in older children and adults!
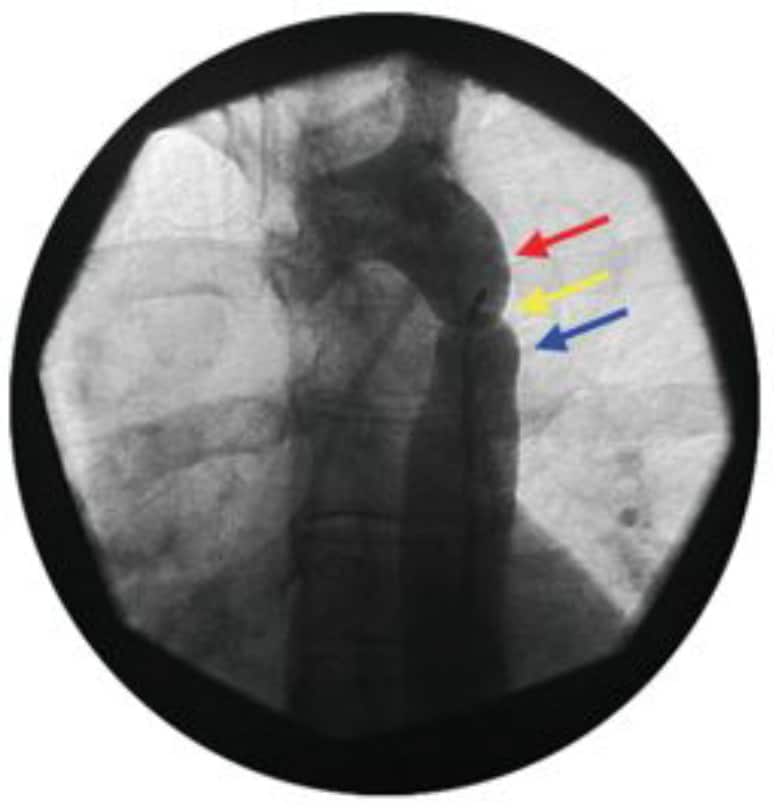
Figure 2. Angiographic view of coarctation of the aorta. The red arrow shows the aortic knuckle, yellow arrow the coarctation and blue arrow poststenotic dilation of the descending aorta.
The combination of the aortic knuckle, coarctation and post-stenotic dilatation forms a characteristic ‘figure 3’ appearance on CXR. Rib notching is seen in older children > 8 years.
Late Presentation – signs and symptoms:
The findings are often more subtle and may relate to associated abnormalities e.g. Turner’s syndrome.
Observation:
- Slightly smaller left arm (if CoA compromises the left subclavian artery)
Palpation:
- Radio-radial or radio-femoral delay
Auscultation:
- CoA murmur – a late systolic murmur posteriorly over the thoracic spine
- Collateral murmurs bilaterally – present in children >5 years due to the formation of collateral arteries from the intercostal vessels. The murmur is crescendo:decrescendo in nature and delayed in onset and termination.
- Evidence of bicuspid aortic valve – either of aortic stenosis (ejection midsystolic murmur); or the early diastolic murmur of aortic regurgitation.
Extracardiac non vascular: features of associated syndromes such as Turner’s or abnormalities of the musculoskeletal system (up to 25% of children with CoA).
INVESTIGATIONS
Blood pressure monitoring
- A difference in blood pressure of 35mmHg between the right arm and lower limbs in older children and adults is characteristic
- Hypertension (take in blood pressure in both limbs)
Non-invasive imaging
Chest X-ray
- Neonate – cardiomegaly, pulmonary oedema
- Child > 8 years and adults – rib notching, prominent aortic knuckle with thoracic indentation (“the figure three sign”; see Fig 2.), cardiomegaly
Echocardiography (transthoracic or transoesophageal)
- May show narrowing across the isthmus.
- The gradient across the CoA can be estimated from blood flow proximal and distal to the CoA.
- Foetal echo may be used to diagnose CoA antenatally but the aorta is technically difficult to image, and the diagnosis frequently inaccurate.
CT
- Rapid acquisition scans are useful to characterise complex anomalies.
MRI
- Useful for the location and extent of CoA as well as other vessel involvement and collaterals. It is also useful for detecting and monitoring aneurysms and re-stenosis post surgery. Data is acquired more rapidly via CT (anaesthesia/sedation frequently required for MRI in children).
Invasive imaging
Angiography
- Useful to measure the peak gradient across the CoA to assess if intervention is necessary (gradient <20mmHg in an older child does not require immediate intervention). It is also valuable for evaluating associated defects and visualising collateral blood flow.
INTERVENTIONS
All patients with a diagnosis of CoA require treatment; the nature and the timing of treatment depends on the severity of the coarctation.
Balloon angioplasty
This is usually reserved for treatment of mild or recurrent CoA in older children. It is not used in infantile CoA as recurrence is common and there is significant risk of injury to the femoral arteries from catheters and aortic rupture during angioplasty.
Surgery
Blalock and Park performed the first end-to-end anastomosis to repair CoA in 1944. This remains the most commonly performed procedure, although a number of different operations have been developed, each with its own advantages and disadvantages. Most surgery is performed via a left thoracotomy between the 3rd and 4th intercostal space. The operation involves cross-clamping the aorta during the repair. The spinal cord is perfused by the anterior spinal artery (arising from the vertebral arteries), and the intercostal arteries that arise from the aorta. Perfusion of the intercostal arteries is lost whilst the aorta is cross cross-clamped, hence spinal cord perfusion is vulnerable during this time. The type of operation depends on surgical preference and is influenced by associated cardiac defects. Figure
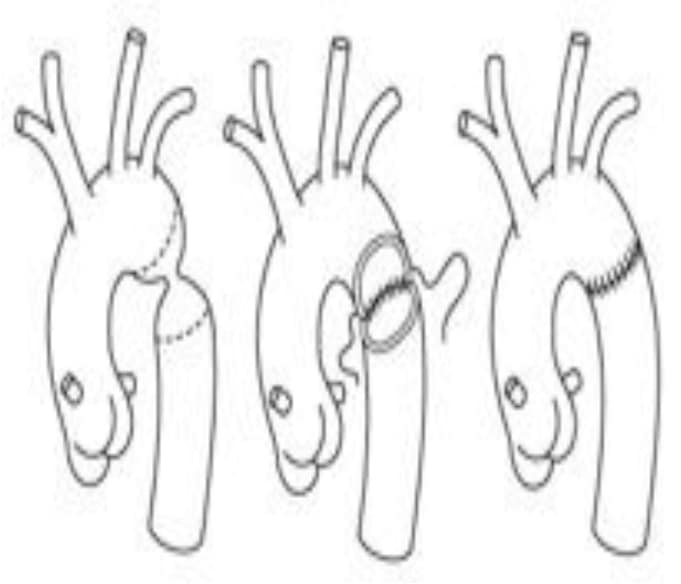
Figure 3. End-to-End anastomosis
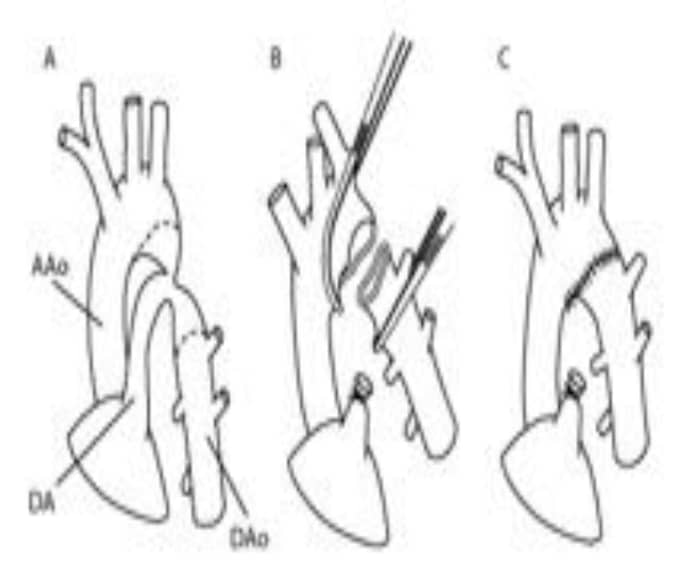
Figure 4. Extended ETE anastomosis
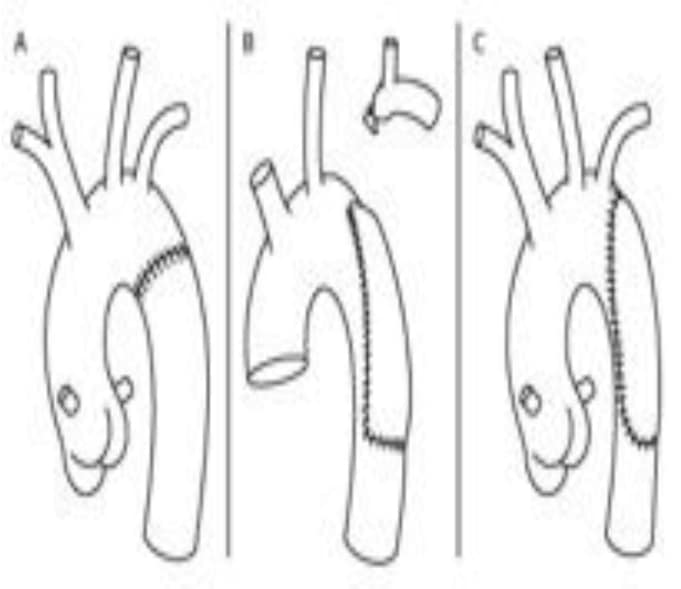
Figure 5. Subclavian flap angioplasty
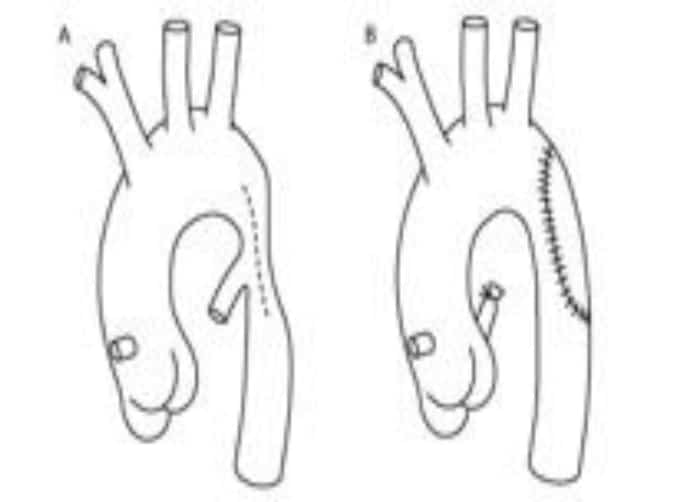
Figure 6. Synthetic patch angioplasty

Table 1: Relative merits of different interventions for coarctation of the aorta
ANAESTHESIA AND PERIOPERATIVE MANAGEMENT
Anaesthetic goals:
Maintain cardiovascular stability, particularly after removal of the aortic cross clamp. Avoid excessive vasodilatation whilst the cross clamp is applied and tolerate relative hypertension during this time. The blood pressure will fall when the aortic cross clamp is removed and coronary perfusion will be jeopardised if hypotension is dramatic. Control hypertension in the postoperative period.
- Effective analgesia using intravenous opioids.
- Minimise the adverse effects of aortic cross-clamp on spinal cord perfusion:
- Maintain MAP >45mmHg distal to clamp
- Surgeon should adjust the proximal and distal clamps to maximise collateral blood flow
- Ventilate to achieve normocarbia in order to minimise cerebral vasoconstriction
- Mild hypothermia (temperature 35°C) to reduce metabolic requirements of spinal cord
- Consider using low dose anticoagulation and monitoring spinal cord function with somatosensory evoked potentials (SSEPs) or motor evoked potentials (MEPs). o Surgeons should minimise cross clamp time to 20 minutes.
Induction:
General anaesthesia may be given using either inhalational or IV agents. Ketamine should be avoided if there is pre-existing hypertension, but is particularly useful in the neonate with duct dependent circulation presenting in extremis.
Monitoring:
Full invasive monitoring, including central and arterial access. A right radial arterial line should be used, with the addition of a femoral arterial line (or blood pressure cuff on the leg) to assess the gradient across the repair. In older children with good collateral supply, a femoral arterial line will suffice. Spinal cord monitoring is used in older children as loss of SSEP/MEP signal is a sensitive indicator of potential spinal cord injury.
Maintenance:
TIVA (not neonates) or volatile (isoflurane is preferred for it’s steal phenomenon which increases blood flow in collaterals, moving blood away from the coarctation) and IPPV.
Analgesia: Intravenous opioids. Thoracic epidurals have been advocated by some for the haemodynamic stability and postoperative analgesia it can provide but many have concerns that this may mask paraplegia in the post operative period as well as enhancing the risk of it occurring. Epidurals should not be used in neonates – local infiltration is preferable.
Postoperative care:
- Aim for extubation at the end of surgery unless complex surgery; monitor in ICU.
- Control blood pressure with sodium nitroprusside infusion in the immediate postoperative period. (3mg/kg SNP in 50ml 5% dextrose (maximum 50mg, at 0.5-4 mcg/kg/min, titrated to effect). Invasive blood pressure monitoring is essential
- β blockers, ACE inhibitors may be introduced postoperatively to control blood pressure
- Good analgesia to minimise hypertension.
- In neonates – introduce enteral feeds carefully as there is a risk of necrotising enterocolitis.
- Monitor lower limb function once muscle relaxants wear off.
COMPLICATIONS
Early complications
Rebound hypertension
This is thought to be due to an increase in sympathetic activity and reflex vasospasm distal to CoA as well as increased renin activity– control with anti hypertensive medication as above.
Post coarctectomy syndrome
This is due to an increase in blood flow and pressure in mesenteric arteries post repair resulting in abdominal distension, pain, vomiting and decreased bowel sounds. This is minimised by aggressive BP control and delaying enteral feeding until day three post op.
Spinal cord injury and paraplegia
This occurs secondary to aortic cross clamping. It is minimised by reducing the duration of aortic cross-clamping and relative hypothermia. In older children SSEP monitoring may be useful. There is an overall incidence of 0.5-1.5%.
Post-operative haemorrhage
This is due to injury to chest wall collaterals seen in older children and adults.
Chylothorax
This is due to damage to lymphatic tissue including damage to the thoracic duct in the vicinity of the repair.
Hoarse voice
This is due to damage of the recurrent laryngeal nerve as it loops around the ductus arteriosus/ligamentum arteriosum.
Diaphragmatic paralysis
This is due to phrenic nerve injury.
Late complications
Hypertension
This occurs in 17-50% of patients. It is exacerbated by exercise, is associated with accelerated cardiovascular mortality and occurs in older patients. It is thought to be due to stiffness in the aorta and altered baroreceptor function as well as increased renin activity and increased extracellular fluid. Cardioselective beta blockers are drugs of choice for treatment.
Re-coarctation and re-stenosis
This occurs in 20% of patients. Contributory patient factors include young age at presentation/repair, tubular hypoplasia of the aorta, a gradient of > 12mmHg across repair and PDA. It also occurs more commonly with a patch angioplasty repair.
SUMMARY
- Coarctation of the aorta is the fifth most common cardiac defect, occurring in 5% of all congenital heart disease
- Mortality approaches 90% if unrepaired by the age of 55.
- Definitive surgical repair is the gold standard
- Paralysis is a rare complication of surgical repair; risk can be minimised by appropriate anaesthetic and surgical measures.
- Hypertension occurs in up to 50% of patients post repair and is associated with accelerated cardiovascular mortality.
ANSWERS TO QUESTIONS
-
- F. Coarctation can occur at any age but the severity determines timing and presentation.
- F. Males are more likely to be affected, the ratio of male: female being 1.7:1 . Caucasians are seven times more likely to get it although the reason remains unclear.
- F. A significant proportion (up to 50%) of patients will experience symptoms such as hypertension.
- T. Mortality approaches 90% by 55yrs of age.
- Early presentation: CoA usually presents in neonates with signs of cardiogenic shock postnatally or after the duct closes depending on the severity and whether there are also other cardiac anomalies. In young children they may present with subtler symptoms of left ventricular dysfunction such as poor feeding, tachypnea, lethargy,failure to thrive and poor exercise tolerance or an incidental finding of upper limb hypertension.
Late Presentation: This usually is asymptomatic and there is an incidental finding of upper limb hypertension or a murmur associated with a bicsupid aortic valve or collateral formaton.The more subtle symptoms may include headache, fatigue and intermittent claudication. Occasionally this may present as a complication of an untreated CoA such as aortic rupture,intracranial haemorrhage, LVF or features of endocarditis. -
- T. This is due to spinal cord ischemia caused by the aortic cross clamp and a good collateral blood supply is thought to be somewhat protective. The incidence varies from 0.5-1.5%.
- F. Hypertension often occurs as an early and late complication/
- F. Haematemesis is not a recognised complication after coarctation repair.
- T. As the thoracic duct empties into the the junction between the left subclavian and left jugular vein , if accidentally damaged, chylothorax can result. Treatment measures include a chest drain and special low fat diet.
- T. This is due to damage to the recurrent laryngeal nerve which loops around the the duct.
REFERENCES and FURTHER READING
- Shah NS. Aortic coarctation. Medscape reference http://emedicine.medscape.com/article/150369- overview (accessed 2nd June 2012)
- May LE, Pediatric Heart Surgery – a ready reference for professionals. Maxishare Milwaukee USA http://www.chw.org/display/PPF/DocID/21357/router.asp (accessed 3rd June 2012)
- Booker PD, Lake CL, Pediatric Cardiac Anaesthesia. Lippincott, Wilkins & Wilkins, 4th edition, 2004, Chapter 21, p443-448.
- Mackay JH, Arrowsmith JE, Core topics in Cardiac Anaesthesia, Cambridge University Press 1st edition, 2004,chapter 43 p232.
- Tsang VT, Stark J, Surgery for Congenital Heart Defects. Wiley, 3rd edition, 2006, Chapter 20, p285- 298.
- Rosenthal E. Coarctation of the aorta from foetus to adult: curable condition or lifelong disease process? Heart 2005; 91: 1495-1502.
- Silversides C, Beauchesne L, Bradley T et al. Canadian Cardiovascular Society 2009 Consensus conference on the management of adults with congenital heart disease: Executive summary Can J Cardiol 2010; 26(3):143-150.
- Hickey EJ, Caldrone CA, McCrindle BW. Left ventricular hypoplasia: A spectrum of disease involving the left ventricular outflow tract, aortic valve and aorta. J Am Coll Cardiol 2012; 59 (sup S): 43-54
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/