Intensive Care Medicine

Self-assessment questions
Question 1
The medical registrar calls you as the anaesthetist on for intensive care. She wants your advice on the management of a 44-year-old woman known to have myasthenia gravis who has been admitted with a cough and pyrexia.
What are the indications for intubation and ventilation?
What treatments are indicated for her myasthenia?
Question 2
You are called to the Emergency Department to assist with a young man with ventilatory failure and profound weakness. He is a college student and has recently returned from travelling, during which time he suffered a diarrhoeal illness. What are the differential diagnoses and what clinical features help make the diagnosis?
Introduction
Patients with muscle weakness may require admission to the ICU because of ventilatory failure or if the patient has suffered, or is at risk of pulmonary aspiration and requires airway protection. In addition, some neurological conditions causing muscle weakness also cause autonomic nervous system failure, requiring invasive monitoring and therapies for haemodynamic support. Significant weakness may also develop during ICU admission – critical illness polyneuropathy and myopathy. This tutorial will discuss the more common disease processes that cause muscle weakness and describe the general management principles for patients with these conditions.
General Management
The requirement for ventilatory support may be prolonged and so nutrition, patient positioning, thromboprophylaxis and other preventative measures are especially important. Consideration must also be given to the inevitable psychological impact on the patient and their family.
The mechanism of ventilatory failure can be sub-divided, but different causes often co-exist:
- (a) Bulbar weakness
Failure of the pharyngeal and laryngeal muscles to maintain airway patency and to protect the airways from aspiration and soiling.
- (b) Inspiratory muscle weakness
Segmental lung collapse leads to reduced functional residual capacity (FRC), atelectasis, infection and ventilation-perfusion (V/Q) mismatch.
- (c) Expiratory muscle weakness
Inadequate cough to clear secretions and open the distal airways, which exacerbates the effects above.
- (d) Complications of immobility
Thromboembolic disease, pneumonia, pressure sores.
The differential diagnosis of the causes of muscle weakness is extensive and a detail description of all conditions cannot be included in this tutorial. The most common causes are described below. ICU management is based on careful attention to monitoring physiological function and early intervention to support failing organ systems, before irreversible damage occurs.
Assessment for ICU admission1 , 2
Regardless of the cause, a systematic approach must be used when assessing and treating a patient with muscle weakness. Timing of intubation and ventilation may be difficult to judge. Impaired conscious level, aspiration, airway obstruction, hypoxaemia or hypercapnoea usually indicate that immediate intervention is needed. Uncertainty about the prognosis and potential for recovery of function in some conditions raises ethical questions about the appropriate level of medical interventions. However most conditions are reversible or controllable and full supportive measures are appropriate. A combination of subjective clinical assessment and lung function tests, in addition to careful consideration of pre-existing physiological reserve and patient wishes, is required to inform management decisions.
Features indicating need for airway protection and ventilatory support are:
- Rapidly progressive weakness
- Difficulty swallowing
- Altered speech
- New onset shortness of breath at rest
- More subjective signs include
- Rapid shallow breathing
- Weak cough
- ‘Abdominal’ breathing – indicating a reliance on diaphragmatic breathing
- Staccato speech – abrupt speech where each syllable is produced separately, indicating poor expiratory function
- Accessory muscle use – abdominal muscles, sternocleidomastoid etc
- Cough or gurgling after swallowing
Lung function tests may not be available and their use is often limited due to poor technique and variability between individuals. Some patients with Guillain-Barré causing facial weakness may be unable to create a seal around the mouth piece. When available, patients lung function tests should ideally be monitored 4 hourly. The tests that are considered most useful include:
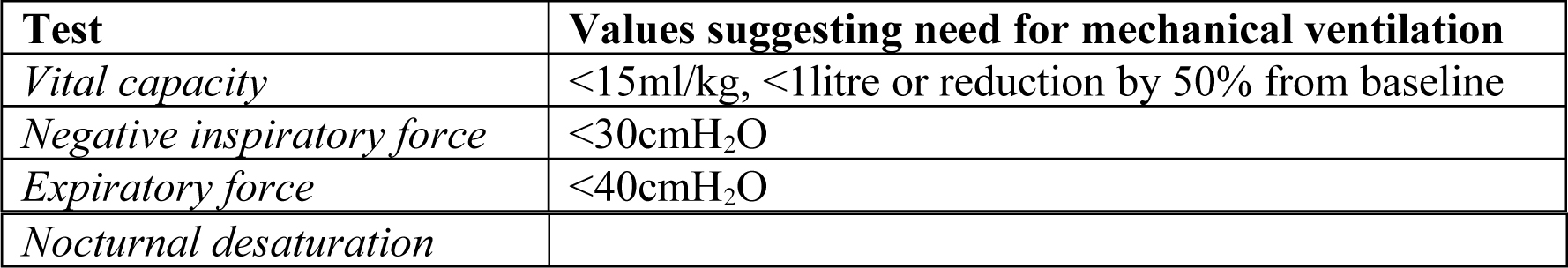
It is vital to be guided by regular clinical reassessment and the trend of PaO2 and PaCO2 on arterial blood gas analysis. Patients with neuromuscular weakness may appear comfortable but be close to decompensation. If intubation is planned in this patient group, previous immobility increases the risk of hyperkalaemia in response to suxamethonium. Consider alternative strategies, such as using rocuronium (9mg/kg) or awake intubation using local anaesthesia.
Neuromuscular causes of weakness
Guillain-Barré Syndrome (GBS)
Epidemiology
GBS is an acute demyelinating polyneuropathy. It is usually (70%) associated with infection, typically Campylobacter jejuni gastroenteritis or respiratory tract infections, but may occur after various other insults such as surgery, vaccination, transplantation and some drugs. Presentation is variable, the classical being an ascending (compared to botulism) flaccid bilateral limb weakness with areflexia. Sensory symptoms are common, including neuropathic pain, but sensory signs are generally absent. It must be suspected in anyone with unexpected limb weakness or sensory deficit. 5-10% of cases will be fatal, with deaths usually due to autonomic nervous system dysfunction, pulmonary embolus or pneumonia. Most patients make a virtually full recovery, but 10 % will not be walking at one year.
Pathophysiology
The aetiology is believed to involve antibody cross-reactivity to components of peripheral nervous tissue and various anti-ganglioside antibodies are found in patients with GBS.
Clinical features
Patients typically present with areflexia, weakness ascending from the lower-limbs and often hyperpathic pain within a month of a potential antecedent factor. Symptoms develop over several days but maybe more rapid, suggesting a worse prognosis. The Miller-Fischer variant presents with ataxia, areflexia and ophthalmoplegia.
Investigations
Specific investigations are useful to guide treatment and include CSF examination, which shows a disproportionate increase in protein levels with respect to CSF leucocytes. There is typically CSF pleocytosis of up to 10 cells/mm3. Pleocytosis of 10-20 cells/mm3 may suggest concurrent HIV infection. Electrophysiological tests demonstrate a pattern of peripheral demyelination.
Treatment
The crucial aspect of management is general supportive measures, potentially for several months. In addition, specific treatment by immunomodulation is effective (see below). Bulbar function can be affected and require definitive airway protection with oro-tracheal intubation or later with tracheostomy. If there has been a period of immobility prior to ventilatory failure, suxamethonium may induce a significant hyperkalaemic response and alternative techniques should be considered.
Autonomic dysfunction in GBS can be very difficult to manage and causes serious morbidity and mortality. Stimulation by laryngoscopy and tracheal suction may precipitate severe cardiovascular instability. Use of topical anaesthetics, judicial use of atropine, where indicated, and small doses of short acting benzodiazepines should be considered. Generally sympathetic activity predominates, with tachycardia and labile blood pressure, but this lability makes treatment difficult and hazardous. If necessary, short acting agents such as esmolol are most appropriate to use. Rarely, bradycardia will require temporary pacing.
Intravenous immunoglobulin (IVIg) and plasmapheresis with albumin replacement are equally effective in improving outcomes, but are of most benefit when started as soon as possible after onset of symptoms.3,4 Plasmapheresis at 50ml/kg/day, 5 times, over 1-2 weeks within 4 weeks of onset increases speed of recovery and improves neurological outcome. There is no benefit to performing more exchanges. Continuous flow plasma exchange machines may be superior to intermittent flow machines and albumin better than fresh frozen plasma as the exchange fluid. IVIg at 400mgkg/day for 5 days is as effective as plasmapheresis, and there is no additional benefit to combining the two therapies.3
The choice between IVIg and plasmapheresis is based on availability and the limits of the patient’s physiology and co-morbidities. IVIg is preferable where the patient has significant haemodynamic instability, sepsis and difficult central vascular access. Plasmapheresis is the treatment of choice when there is concurrent renal failure, congestive heart failure, hyperviscosity or IgA deficiency (risk of anaphylactic reaction with IVIg). Corticosteroids have been shown to be ineffective and in some studies have been associated with a worse outcome. CSF filtration may be of benefit but is currently considered an experimental treatment.
GBS does not affect conscious level, and the prolonged course will inevitably have psychological consequences. Sedation should be used when necessary, though oversedation for prolonged periods is likely to worsen psychological as well as physical recovery. Occupational therapy and physiotherapy are of importance in rehabilitation and should start as soon as is practicable. Passive exercise is important to reduce muscle wasting and prevent contractures.
Prognosis
GBS has a mortality of 5-10% and 10% will not be walking after a year.
Myasthenia Gravis (MG)
Epidemiology
MG has an annual incidence of about 5 per million population and MG prevalence is about 10 per 100 000 population. In young Caucasian adults, most cases are female, with a shift to males in the over 50s. Pre-pubertal presentation is relatively common in Asians.
Pathophysiology
MG is an autoimmune disease, characterised by production of an autoantibody against the post-junctional nicotinic acetylcholine (ACh) receptors of the neuromuscular junction. Receptor numbers are greatly depleted, resulting in a characteristic fluctuating weakness with fatigability of bulbar, ventilatory, extra-ocular and proximal upper limb muscles. Reflexes, sensation or other neurological functions are usually intact.
Clinical features
MG should be suspected in any patient with weakness associated with fatigability, especially in the presence of ptosis, diplopia, poor head position control, flaccid dysarthria (nasal, staccato speech), chewing weakness and difficulty swallowing. An uncommon sub-type, characterised by the presence of muscle-specific tyrosine kinase antibodies, may present with ventilatory failure. Motor weakness should improve with rest and increase significantly with administration of a cholinesterase inhibitor such as edrophonium or neostigmine. Deep tendon reflexes are present, in contrast to GBS. The combination of one or more of the above and one or more of the following features confirms the diagnosis:
- ACh receptor antibodies
- Characteristic electromyographic (EMG) studies – increased single fibre ‘jitter’, decremental response to repetitive peripheral nerve stimulation, reversed by a cholinesterase inhibitor.
Exacerbating factors
With known cases of MG it is important to identify, avoid or minimise factors that may precipitate a myasthenic crisis.
- a. Lower respiratory tract infections (probably most common)
- b. Aspiration / airway soiling
- c. Sepsis
- d. Surgery
- e. Reduction of immunological therapy
- f. Commencement of steroid therapy
- g. Pregnancy
- h. Drugs:
- i. Neuromuscular blocking drugs – profoundly sensitive to non-depolarising agents, probably resistant to depolarising agents (suxamethonium)
- j. Some antibiotics – aminoglycosides especially gentamicin, macrolides
- k. Beta-blockers, calcium channel blockers, procainamide, quinidine
- l. Quinine
- m. Corticosteroids
- n. Magnesium
- o. Tocolytics
- p. Iodinated contrast agents
- q. Penicillamine
Treatment
The most commonly used cholinesterase inhibitor is pyridostigmine. The dose for adults starts at 30mg 4-5 times a day, up to a maximum of 60mg 4-5 times a day. Higher doses run the risk of precipitating a cholinergic crisis, which causes weakness through depolarization neuromuscular block and may also result in ventilatory failure. The characteristic features of a cholinergic crisis allow it to be clinically differentiated from a myasthenic crisis and include:
- Hypersalivation, lacrimation and sweating
- Miosis (constricted pupils)
- Abdominal pain, nausea, diarrhoea, vomiting
- Bradycardia
Any concern about cholinergic excess should be managed by intubation and ventilation. Once intubated, anticholinesterases should be stopped to reduce cholinergic complications.
In myasthenic crisis plasmapheresis is effective in improving muscle power, although it may not improve functional outcome5. There is less evidence for the use of IVIg for moderate to severe MG,6 however some authorities recommend its use if plasmapheresis is contraindicated or unavailable.
Patients presenting with a crisis may have had significant doses of steroids and other immuno-suppressants. Thymectomy is a well-established treatment for certain subgroups – this is specialist surgery, requiring careful pre-operative planning and optimization of muscle function. A patient in myasthenic crisis is unlikely to benefit from thymectomy acutely.
Non-invasive ventilation (NIV) may be considered as a temporizing measure, as long as the airway is patent and enough bulbar function remains to clear secretions. Patients with MG may do better with NIV than others with neuromuscular weakness, as it provides muscle rest and allows strength to improve. Many progress to need intubation and invasive ventilation.
Prognosis
The general outlook is good, with 90 per cent achieving near-normal functional recovery. The side effects of potent immunomodulatory treatment may significantly impact on mortality and morbidity.
Botulism
Epidemiology
Botulism is rare in the developed world. The first recorded cases of food-borne botulism in the UK occurred in 1922, caused by duck paste sandwiches. Eight people were affected and all died. Ten incidents have been reported since, with 11 deaths among the 50 people concerned. Five of the 11 were caused by commercially produced foods. No single food or type of food has predominated; five were vegetarian, four meat, and two fish. Classic food-related botulism is rare, but in recent years, an increase in wound botulism associated with injected drugs (particularly black tar heroin) has been seen.
Pathophysiology
Toxins formed by the micro-organism Clostridium botulinum interrupt neuromuscular transmission by cleaving proteins necessary for release of acetylcholine from nerve terminals. This also affects transmission at autonomic ganglia and parasympathetic nerve terminals. The process is permanent. The toxins are either ingested pre-formed (e.g. food poisoning) or formed in vivo (e.g. wound, infant and adult intestinal botulism). The use of botulinum toxin as a biological weapon results in inhalational botulism.
Clinical Features
Signs and symptoms of food-borne botulism generally develop within 12-36 hours of ingestion of contaminated food. The severity is proportional to the amount of toxin ingested. The clinical picture is a rapid onset symmetrical descending flaccid paralysis, with multiple cranial neuropathies, in the absence of fever or altered consciousness. Gastrointestinal symptoms including nausea, vomiting, diarrhoea and colicky pain, may precede the neurological signs. These are absent in wound botulism, which has a longer incubation period of up to a week. Parasympathetic dysfunction may present early, with dry mouth and blurred vision associated with dilated, poorly reactive pupils. Further autonomic dysfunction may manifest as gastrointestinal dysmotility, orthostatic hypotension, altered resting pulse, urinary retention, or hypothermia. Diplopia often develops secondary to extraocular muscle weakness. Bulbar weakness may result in flaccid dysarthria, chewing difficulty, and dysphagia. The upper limbs, trunk and lower limbs may become weak in a descending pattern. Respiratory compromise occurs due to a combination of upper airway obstruction from weak oropharyngeal muscles and diaphragmatic weakness.
Treatment
Prolonged (30-60 days) ventilatory support is usually necessary, as recovery depends on re-growth of nerve terminals. Trivalent antitoxin may reduce severity, but due to the irreversible binding of the toxin, it must be given as early as possible.
Prognosis
With improvements in respiratory care, the case-fatality rate has improved from 60% during 1899 to 1949, to 12.5% during 1950 to 1996. The fatality risk for the index case in an outbreak is 25%, with a 4% fatality risk for subsequent cases after recognition of an outbreak. The public health implications of botulism make it mandatory to report it to the relevant public health body.
Muscle weakness acquired during critical illness
Weakness acquired during intensive care may be due to GBS, unmasked myasthenic disorders, spinal cord infarction or electrolyte imbalance. However, it is now recognised that the most common muscular causes of failure to wean from mechanical ventilation in ICU are critical illness polyneuropathy, critical illness myopathy and prolonged neuromuscular blockade.
Electrolyte disorders
Disturbance of biochemical homeostasis is common in the intensive care unit and the principle electrolyte abnormalities contributing to muscle weakness and ventilatory failure are:
- Hypermagnesaemia
- Hypophosphataemia
- Hypo/hyperkalaemia
Rhabdomyolysis
This potentially devastating cause of muscle weakness must be considered and excluded. Causes include various drugs, trauma, surgery and prolonged muscle compression.
Critical illness polyneuropathy – CIP
Initially described in the early 1980’s, this condition is associated with the systemic inflammatory response syndrome (SIRS), sepsis and multiple organ failure. 70% of such patients will have electrophysiological features of CIP and 30% will have subsequent difficulty weaning from ventilation. It is a widespread axonal peripheral neuropathy causing weakness and wasting of extremity muscles, distal sensory loss and paraesthesia. The cranial nerves are typically spared. The main differential diagnosis is GBS, which is often excluded on clinical grounds, with electrophysiological studies rarely being necessary.
The prognosis will depend on resolution of the antecedent disease, but survivors should recover good function over several months. CIP does not seem to adversely affect long-term survival.
Recently, tight glycaemic control has been shown to reduce the development of CIP.
Critical illness myopathy – CIM (Acute myopathic quadraplegia)
This condition is typically associated with severe respiratory disease, such as status asthmaticus. Most cases occur after use of non-depolarising neuromuscular blocking drugs and high dose corticosteroids, although cases also occur independently of these factors. Many other predisposing agents have been identified, such as muscle relaxation induced by other drugs e.g. propfol or benzodiazepines.
A flaccid global weakness develops after several days of muscle relaxation but sensation remains intact. Serum creatine kinase may be elevated especially if measured early in the course of the myopathy. Electrophysiology studies demonstrate normal sensory action potentials, but reduced amplitude of compound muscle action potentials. Muscle histology, which is unnecessary for clinical management, shows myosin loss. Like CIP, CIM may require prolonged ventilatory support, but should not worsen the patient’s long-term outcome, with muscle function typically recovering over weeks to months.
Where possible the use of high doses of corticosteroids should be avoided, especially when non-depolarising muscle relaxants are being administered. There is little to be gained from differentiation between these two conditions, as management is the same for both. Some authors feel that they represent two ends of a spectrum of disease.
Prolonged Neuromuscular Blockade
Continuous infusions of neuromuscular blocking agents have been associated with delayed return of muscle strength. This is due to the effects of hepatic and renal dysfunction on the metabolism of steroidal muscle relaxants such as vecuronium. This problem can be significantly reduced by limiting the use of these agents through appropriate ventilator modes, available on more modern machines and through effective, appropriate sedation. If absolutely essential, infusions should be monitored properly by peripheral nerve stimulation and adjusted to maintain a minimal level of muscle relaxation i.e. 1-2 twitches in response to supra-maximal stimuli in a train-offour pattern. Alternatively, daily infusion breaks, allowing return of muscle activity before re-paralysing, may be useful.
References and further reading
- Mehta S. Neuromuscular disease causing acute respiratory failure. Respir Care 2006;51(9):1016 -21; discussion 1021-3.
- Dhand UK. Clinical approach to the weak patient in the intensive care unit. Respir Care 2006;51(9):1024-40; discussion 1040-1.
- Hughes RA, Raphael JC, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barre syndrome. Cochrane Database Syst Rev 2006(1):CD002063.
- Hughes RA, Swan AV, van Koningsveld R, van Doorn PA. Corticosteroids for Guillain-Barre syndrome. Cochrane Database Syst Rev 2006(2):CD001446. e.
- Gajdos P, Chevret S, Toyka K. Plasma exchange for myasthenia gravis. Cochrane Database Syst Rev 2002(4):CD002275.
- Gajdos P, Chevret S, Toyka K. Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst Rev 2006(2):CD002277.
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/