Paediatric Anaesthesia

KEY POINTS
- Patients with metabolic bone disease may present for surgery unrelated to their disease and to nonspecialist centres.
- The symptoms and signs of metabolic bone disease can vary widely between individuals, including those with the same disease, necessitating an individualised risk-management approach.
- Patients can be at increased risk during perioperative care including airway difficulty, fracture and bleeding.
- Electrolytes including calcium and phosphate may be deranged and should be checked prior to surgery when relevant.
- Where possible, involvement of specialist teams including metabolic bone, cardiology, intensive care and pain medicine can reduce perioperative risk.
INTRODUCTION
Bone is a metabolically active organ comprising matrix, minerals and cells. The interaction of these components is influenced by multiple factors, including hormones. Disorders in any of the components can result in metabolic bone disease (MBD).
This article intends to summarise the most relevant forms of paediatric MBD and their implications for perioperative care. Children may present for surgery as part of their treatment or for unrelated surgery, at either specialist or nonspecialist centres. Practitioners providing anaesthetic care should have a working knowledge of the clinical features of this group of disorders and take steps to minimise risk during surgery.
Metabolic Pathways
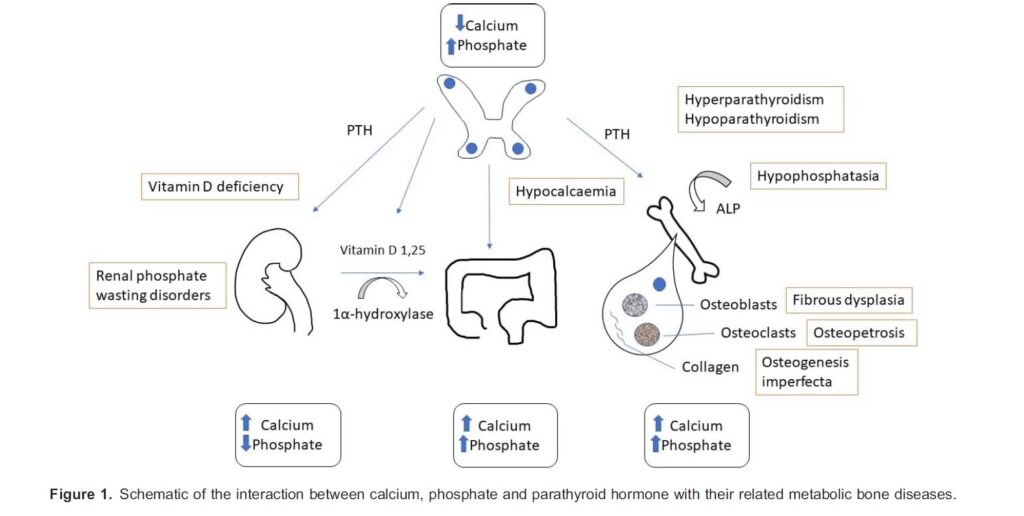
Several ions, molecules, hormones and enzymes are involved with the metabolism of bone. In response to low plasma calcium, the parathyroid glands produce parathyroid hormone (PTH). This hormone acts on the kidney to increase reabsorption of urinary calcium and also via the production of 1,25 dihydroxyvitamin D on the gut to increase the absorption of calcium and phosphate. Osteoblasts are the cells that make bone tissue (osteoid) and enable its mineralisation. PTH stimulates osteoclasts, the cells that resorb bone mineral, leading to increased serum calcium and phosphate.
Vitamin D plays a crucial role. In the skin, in response to UV-B light, cholecalciferol (pre–vitamin D3) is synthesised from 7- dehydrocholesterol. The dietary source of vitamin D is ergocalciferol (vitamin D2). After absorption in the gut, ergocalciferol undergoes hydroxylation in the liver to produce 25-hydroxyvitamin D (calcidiol). Calcidiol undergoes further hydroxylation in the renal tubule by the action of PTH to produce 1,25-hydroxyvitamin D or calcitriol.1
DISORDERS OF BONE MINERAL ION HOMEOSTASIS
Mineral in bone is mainly in the form of hydroxyapatite, which contains calcium and phosphate. (Figure 1)
Rickets
Rickets is a term used when there is failure of mineralisation of growing bones. This is characterised by typical radiographic appearances, poor growth, skeletal deformity and sometimes bone pain. There are many causes of rickets; the most common is acquired vitamin D deficiency and lack of dietary calcium (calciopenic), the clinical effects of which are largely preventable with detection and treatment.1 Rickets due to a lack of phosphate (phosphopenic rickets) is most commonly the result of an inherited disorder and is less common than calciopenic rickets.
X-Linked Hypophosphataemic Rickets
The most common genetic cause of rickets is X-linked hypophosphataemic rickets (XLH). This disorder results from mutations in the PHEX gene (Xp22.1) and is transmitted as an X-linked dominant trait. The PHEX gene regulates a protein called fibroblast growth factor 23 (FGF23), a protein that effectively inhibits the renal reabsorption of phosphate. The mutated gene leads to the increased production of FGF23 and thus reduced phosphate reabsorption in the kidney, resulting in hypophosphataemia.1,2
Incidence
XLH has a prevalence of approximately 1 in 20 000. The male-to-female ratio is 1:1.
Clinical Features
Clinical features of XLH include poor linear growth with bowing of the legs around walking age, bone pain, craniosynostosis (premature closure of the skull sutures) and dental abscesses. Biochemical features include hypophosphataemia and raised alkaline phosphatase, typically with normal calcium, parathyroid hormone and vitamin D concentrations.
Surgery
Surgery is usually unrelated to the patient’s bone disease. Some patients require orthopaedic intervention to correct skeletal deformity and neurosurgery to release craniosynostosis.
Bone Disease of Prematurity
Bone disease or osteopenia of prematurity are terms used to indicate the inadequate mineralisation of the skeleton of premature neonates driven by the failure to provide sufficient mineral to meet the needs of growing bone. It is more common in babies born at less than 28 weeks gestation, as most bone mineralisation takes place in the third trimester of pregnancy.3 As a result, this condition should be considered in all premature neonates presenting for surgery, particularly those who have been critically ill.
Clinical Features
Bones are prone to fractures (risk factors include ,28 weeks of gestation, chronic lung disease, steroid and diuretic use and conjugated hyperbilirubinaemia).4 There may be classic radiographic features of rickets.3 Undermineralisation of the rib cage can predispose to rib fractures and/or insufficient rigidity to adequately support spontaneous ventilation, with the potential for cardiorespiratory sequalae (e.g., difficulty weaning from mechanical ventilation). There is secondary hyperparathyroidism (see below), which responds to adequate mineral ion replacement. Sometimes vertebral fractures can be seen in more severe cases of bone disease of prematurity.
Surgery
Surgery is generally performed in this patient age group only to treat life-threatening disease (eg, laparotomy). However, when performing a surgical procedure, an awareness of potential bone fragility and undermineralisation is important.
Hyperparathyroidism
Primary hyperparathyroidism is an uncommon childhood disease whereby excess PTH is produced, either from parathyroid gland adenomas, glandular hyperplasia or, very rarely, malignancies. The PTH acts on bone and the kidney to increase the reabsorption of calcium, resulting in hypercalcaemia. Secondary hyperparathyroidism is more common and results from chronic hypocalcaemia (eg, from vitamin D deficiency or chronic kidney disease).
Clinical Features
Mild hypercalcaemia is often asymptomatic. As calcium levels increase, symptoms in older children include thirst, polyuria, abdominal pain, constipation, confusion, renal stones and potentially renal failure. In infants, hypotonia, poor feeding and irritability are reported. Rarely, infants can present with coarse cry from vocal cord paralysis and in extremis with respiratory failure resulting from muscle weakness and rib fractures (eg, in neonatal severe hyperparathyroidism).2
Surgery
Rarely, children may present for parathyroidectomy. More commonly, patients present for renal stone surgery, diagnostic procedures or unrelated surgery.
DISORDERS OF BONE MATRIX
The bone matrix is made up of collagen fibres and a network of interacting proteins and other molecules. Type 1 collagen is the most abundant form in bone, constituting 90% of the nonmineralised component.
Osteogenesis Imperfecta
Osteogenesis imperfecta (OI) is a rare inherited disorder of type 1 collagen production and/or processing. Inheritance can be autosomal dominant, autosomal recessive or result from sporadic mutations.5 There are other less common causes of primary osteoporosis.
Incidence
Approximately 1 in 20 000 births are affected by OI. The male-to-female ratio is 1:1.
Background
Like many other inherited metabolic disorders, OI is a spectrum of disease. Classically, there have been 4 subtypes described,5 although more recent classifications list as many as 20 subtypes,6 with a range of clinical manifestations and significant overlap of symptoms. All subtypes have soft and weak bone that is prone to fracture. Patients may also have dentinogenesis imperfecta (discoloured and weak teeth). Some subtypes have extra-axial manifestations including aortic root dilatation, valvular heart defects, coagulopathy and deafness. Patients are often of short stature and may have significant skeletal deformity including kyphoscoliosis.
Surgery
Surgical treatment includes fracture fixation, intramedullary rods (most commonly long bone rodding in type 3) and deformity correction. Spinal surgery for scoliosis and, rarely, skull base surgery are performed to stabilise basilar invagination. Patients may also present for dental surgery.
Secondary Osteoporosis
This common adult disease can present in childhood and is associated with long-term steroid therapy, inadequate nutrition, inflammatory disorders and chronic immobility.
OTHER DISORDERS
Hypophosphatasia
Hypophosphatasia (HPP) is a rare inherited disorder of defective mineralisation characterised by the poor mineralisation of bones and teeth.
Incidence
The incidence of HPP is approximately 1:100 000 live births. Males and females are equally affected. There is a higher incidence of HPP in certain ethic groups (1:2500 Canadian Mennonites).5
Background
HPP is caused by the mutation of the tissue-nonspecific alkaline phosphatase (ALPL) gene leading to reduced alkaline phosphatase levels. There are 6 major clinical subtypes of HPP, ranging from a severe form that can lead to in utero or neonatal death to a form associated with only premature loss of deciduous teeth without discernible bone abnormalities. Generally, more severe subtypes of HPP are associated with an earlier age of onset, and severity correlates with the amount of residual alkaline phosphatase activity in the body, with less enzyme activity resulting in more severe disease.7
Clinical Effects
There is wide variability between individuals, even amongst members of the same family. Bones can be weak and prone to fracture and deformity. Infants and young children may have craniosynostosis; the resulting skull abnormalities may also lead to intracranial hypertension. In severe disease, deformities of the thorax and rib fractures predispose infants to pneumonia and respiratory compromise. Other problems in severe infantile disease include hypercalcaemia and nephrocalcinosis. Older children may have bone pain, fatigue and delayed motor development.
Surgery
Surgical treatment includes fracture management and deformity correction. Surgery may also be performed for craniosynostosis.
Osteopetrosis
This rare disease results from mutations in genes responsible for osteoclast function or quantity. Defective bone resorption ensues, resulting in skeletal and metabolic effects. There is increased bone density, but the poor quality of bone renders it prone to fracture. There are several described causes of osteopetrosis,2 classically described as benign, intermediate or malignant and distinguished by clinical manifestations and inheritance pattern.8 Malignant forms carry high mortality rates without hematopoietic stem cell transplantation.9
Incidence
Benign types have an incidence of ~1 in 20 000. Malignant forms are rare, with an estimated incidence of 1 in 250 000.
Clinical Features
Benign types present with bone pain and fractures, deformity including scoliosis, short stature and impaired growth. In addition, patients with malignant forms may have the following:
- Cranial nerve impingement: hearing/visual loss, facial nerve palsy
- Disordered bone marrow: anaemia, thrombocytopenia and impaired immunity (increased risk of osteomyelitis)7
- Bony overgrowth: choanal stenosis, restricted mouth opening and reduced neck extension (cervico-medullary stenosis)
- Hypocalcaemia
Surgery
Surgery includes fracture fixation, dental procedures and vascular access for stem cell transplantation or antibiotics to manage osteomyelitis. Bone marrow aspiration may be performed prior to and following hematopoietic stem cell transplantation.
Fibrous Dysplasia and McCune-Albright Syndrome
Fibrodysplasia (FD) is a rare sporadic MBD in which normal cancellous bone is replaced by fibrous tissue due to a somatic mutation in the GNAS1 gene. The resulting abnormal bone can be painful and prone to pathological fracture.10 It is commonly monostotic (isolated to one bone) but can be polystotic (affecting multiple sites). Rarely, it occurs as part of a syndrome, termed McCune-Albright syndrome (triad of FD with cafe´ -au-lait spots and hyperfunctioning endocrine tissue). The condition can also be associated with hypophosphataemia due to elevated levels of FGF23 (see above).
Incidence
The exact incidence of FD is not known. McCune-Albright syndrome has an estimated incidence of 1 in 100 000 to 1 in 1 000 000 people worldwide.
Clinical Features
Symptoms vary according to the site affected, and lesions can be asymptomatic. The most commonly affected areas are the long bones of the legs (eg, ‘‘shepherd’s crook’’ deformity of proximal femur), ribs, skull and facial bones. Bone pain, fractures, deformity, asymmetrical growth including leg length discrepancy and scoliosis may be present. Facial deformity (asymmetry) can feature; therefore, careful airway assessment is advised. Visual disturbance and hearing loss can also occur. Skull base disease is a risk factor for visual loss. As growth hormone excess may be a driver of skull base disease, some children are on inhibitors of growth hormone release (eg, lanreotide). Depending on the extent and severity of disease, patients with McCune-Albright syndrome may also have cardiac problems, thyroid disturbance and electrolyte abnormalities causing arrhythmias, and liver disease.10
Surgery
Patients may require surgery to correct skeletal deformity and rarely to decompress neurologic structures including ophthalmic and neurosurgical intervention. More commonly required procedures are fracture fixation and instrumented spinal fusion.
ANAESTHETIC CONSIDERATIONS
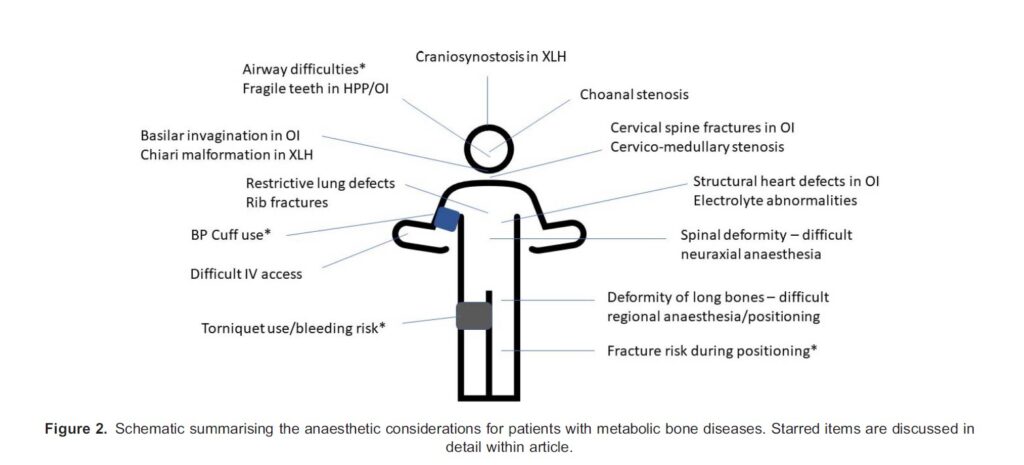
If the child has had previous anaesthesia, it is prudent to review these records, paying particular attention to airway interventions, intravenous access, ventilation, positioning, monitoring used and any complications or difficulties reported. Note the size, proportions and weight including weight distribution of the patient, relative to their age and peers. (Figure 2)
Airway Difficulties
In our experience, most patients with MBDs have airways that can be managed without particular difficulty. Some clinical features can lead to difficult laryngoscopy and intubation including the following:
- Malocclusion from mandibular hypoplasia or bony overgrowth
- Reduced temporomandibular joint function and restrictive mouth opening
- Choanal stenosis (precluding nasal intubation, additional care on extubation)
- Reduced cervical spine movement or the desire to maintain its neutral position
- Decreased tolerance of apnoea owing to VQ mismatch from thoracic deformity
Endotracheal tube size is generally that expected for age, as the laryngeal development correlates with this. Supraglottic airway devices, however, are more likely to be chosen according to weight and height.
Mineral Ions
Urea and electrolytes, Ca, Mg, PO4 should be checked preoperatively unless previously normal with no further disease progression.
Calcium
Treat hypocalcaemia with calcium replacement. Beware the risk of seizures and arrythmias if hypocalcaemia is uncorrected. Hypercalcemia leads to systemic vasoconstriction with contraction of the intravascular space; volume expansion with intravenous crystalloid may be required.
Magnesium
Arrhythmias are more likely if the magnesium level is low. If overtreated, neuromuscular weakness may occur, and the action of muscle relaxants may be prolonged.
Phosphate
Hypophosphataemia may be seen in renal-phosphate wasting disorders (XLH, autosomonal dominant hypophosphataemic rickets, Fanconi syndrome) and vitamin D resistance. Untreated hypophosphataemia has multisystemic effects including muscle weakness, respiratory failure and cardiac arrhythmias.11
Fracture Risk
Establish the severity of their disease and note previous fractures.
As described above, patients with some types of MBD are at higher risk of sustaining fractures during handling and positioning, including when placing intravenous lines and applying monitoring. Treatment with bisphosphonates has reduced the incidence and risk of fractures and radically altered patient outcomes for this group of patients. A recent case series at a tertiary centre found that the incidence of perioperative fractures in patients with OI was low,12 demonstrating that with careful handling, this risk can be managed.
However, extreme caution is still required. The need for transferring anaesthetised patients with MBD should be minimised. When unavoidable, it is essential that theatre staff are briefed regarding the risks to the patient, communicate clearly and handle patients with additional caution. Check and protect pressure points with pressure-relieving padding. Clear sterile drapes allow the patient to be visible during surgery, which may reduce the risk of pressure injury from surgical instruments or personnel. Counsel patients and their families about these risks and the steps that you will take to mitigate them.
Consider the use of a sedative premedicant to reduce the likelihood of sudden movements or the requirement for restraint. Whilst inhalation inductions may be helpful for patients with difficult intravenous access, they may risk injury to the patient during the excitation plane of anaesthesia. This must be balanced against alternative options but can be carried out with care.
Tourniquets
Classically, the use of tourniquets in patients with MBD is contraindicated owing to concerns regarding increased fracture risk. However, some centres experienced in their use, including our own, have not reported an increased fracture risk. Patients with OI type 3 have a higher risk of perioperative haemorrhage,12 and the decision to use tourniquets must therefore be balanced.
Blood Pressure Cuffs
Blood pressure (BP) cuffs should be used with caution in patients with some MBDs. If required, place the BP cuff over an area that has had intramedullary rods or refer to previous theatre documentation. The BP cuff may need to be cycled less frequently. Alternatively, consider arterial cannulation and invasive BP monitoring in patients in whom blood loss or fluid shifts are anticipated.
Other Considerations
Temperature Complications
Hyperthermia has been reported in patients with OI. There is mixed evidence in the literature, but the suggestion is that this is not malignant hyperpyrexia.13 In our experience, patients with severe OI often become hyperthermic during anaesthesia. On the contrary, patients with MBD may be of short stature and have decreased muscle mass. They are frequently exposed during
procedures, and surgical skin prep will contribute to heat loss. These patients are at high risk of perioperative hypothermia. Continuous temperature monitoring is desirable in these patients perioperatively.
Bleeding Diathesis
Patients with OI have an increased risk of bleeding.12 Check clotting preoperatively, use blood conservation strategies, encourage open channels of communication between the surgical and anaesthetic team and liaise closely with blood bank and haematology. Thrombocytopenia is associated with osteopetrosis. Check platelet count preoperatively and prior to regional anaesthesia. In our institution, we routinely administer tranexamic acid to OI patients undergoing surgery with any risk of excessive bleeding.
Drug Dosing
In our experience, drug doses should be calculated based on the weight of the patient, with caution at extremes of weight.
Regional Anaesthesia
Patients with MBD often benefit from regional anaesthesia (RA) techniques during orthopaedic surgery. In our centre, we find that this can be carried out safely and is an effective opioid- paring technique. Positioning for RA may be more difficult due to deformity, and we advocate the use of ultrasound to avoid both fractures from nerve stimulation and confirm the anatomy. Patients and their carers need to be counselled not just about the usual risks of RA but also those risks specific to their condition and block. The reduced sensation postoperatively will necessitate increased caution when moving or mobilising.
Intraosseous Access
Intraosseous access is relatively contraindicated in patients with bone fragility disorders owing to the increased fracture risk. In the general population, however, iatrogenic fracture from intraosseous access is rare.14
General Considerations
Communication
Patients may have hearing or visual impairment and, as such, experience difficulties in communicating effectively in hospital. Patients may be institutionalised and may have a good understanding of how they would like their care to be given. Pay attention and ensure that staff use age-appropriate communication, as patients may appear younger due to their size.
Chronic Pain
Some patients with MBD are affected by chronic pain. Consider this during the preoperative assessment and use strategies to manage this effectively. Where available, the involvement of a pain specialist during perioperative care is recommended.
SUMMARY
Whilst primary bone diseases in childhood are generally rare, collectively they constitute a set of diseases that are likely to be encountered by a paediatric anaesthetist. In addition, there is a broad range of conditions affected by secondary bone disease, in which similar concerns regarding bone fragility commonly apply (eg, in premature infants). The importance of an awareness of the implications of bone disease is highlighted when one considers the range and
potential severity of complications.
REFERENCES
1. Elder C J, Bishop N J. Rickets. Lancet. 2014;383:1665-1676.
2. Allgrove J. Metabolic bone disease. Paediatr Child Health. 2010;21(4):187-193.
3. Greer FR. Osteopenia of prematurity. Annu Rev Nutr. 1994;14:169-185.
4. Bishop N, Sprigg A, Dalton A. Unexplained fractures in infancy: looking for fragile bones. Arch Dis Child 2007;92:251-256.
5. Bissonnette B, Luginbuehl I, Marciniak B, et al. Syndromes: Rapid Recognition and Perioperative Implications. 1st ed. New York: McGraw-Hill Medical; 2006.
6. Chetty M, Roomaney IA, Beighton P. The evolution of the nosology of osteogenesis imperfecta. Clin Genet. 2021;99(1):42- 52.
7. Whyte MP. Hypophosphatasia—aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2016;12:233-246.
8. Tolar J, Teitelbaum SL, Orchard PJ. Osteopetrosis. N Engl J Med. 2004;351(27):2839-2849. 9. Burgoyne LL, Kaur A, Billups CA et al. Complications of anesthesia for children with malignant infantile osteopetrosis before and after hematopoietic stem cell transplantation. Pediatr Anesth. 2010;20:1046-1051.
10. Javaid MK, Boyce A, Appelman-Dijkstra N, et al. Best practice management guidelines for fibrous dysplasia/McCune- Albright syndrome: a consensus statement from the FD/MAS international consortium. Orphanet J Rare Dis 2019;14(1):139.
11. Wadsworth RL, Siddiqui S. Phosphate homeostasis in critical care. BJA Educ. 2016;16(9):305-309.
12. Rothschild L, Goeller JK, Voronov P, et al. Anesthesia in children with osteogenesis imperfecta: retrospective chart review of 83 patients and 205 anesthetics over 7 years. Pediatr Anesth. 2018;28:1050-1058.
13. Rothschild L. Best practices for using anesthesia in patients with OI. Osteogenesis Imperfecta Foundation Podcast Series. Accessed January 24, 2019. https://oif.org/informationcenter/podcast/
14. Paxton J. Intraosseous vascular access: a review. Trauma. 2012;14(3):195-232.
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/