Intensive Care Medicine

KEY POINTS
- Phosphate is key to many physiological processes.
- Average dietary phosphate uptake is 1 to 5 g (20-40 mmol) per day.
- Refeeding syndrome is a potentially lethal complication of prolonged hypophosphataemia that requires early detection and prompt treatment.
- Hyperphosphataemia (serum phosphate concentration >5 mmol/L) can lead to dangerous hypocalcaemia and precipitation phenomena.
INTRODUCTION
Phosphate is a key electrolyte with a variety of roles in the body. Phosphate derangement is a marker of illness severity and is often found as part of a concurrent illness, either from an acute change or a protracted deficit such as in chronic alcoholism or starvation.1,2 Phosphate derangement may also be associated with increased mortality; however, there are several conflicting studies, so this is not established fact.1,3
Phosphate derangement is common in the intensive care unit (ICU) population, with about 30% to 80% of ICU patients having hypophosphataemia and up to 45% having hyperphosphataemia, compared with 2% to 3% of the general inpatient hospital population.3 It is imperative to consider underlying mechanisms when correcting phosphate derangement, as this will provide patient context, guide correction and avoid overcorrection or inadvertent hypocalcaemic states. This tutorial will focus on phosphate physiology and homeostasis, mechanisms of hypophosphataemia and hyperphosphatemia, clinical presentation and management protocols relevant to the ICU.
PHOSPHATE
The Physiological Roles of Phosphate
Phosphate has a role in essential processes throughout the body, with uptake, storage and excretion regulated by several mechanisms (Figure 1). The normal range for plasma phosphate concentration is defined as 0.8 to 1.45 mmol/L.4
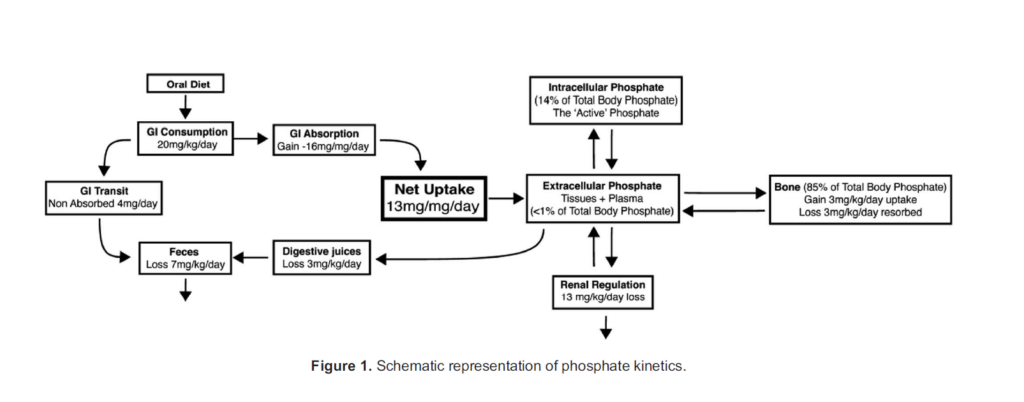
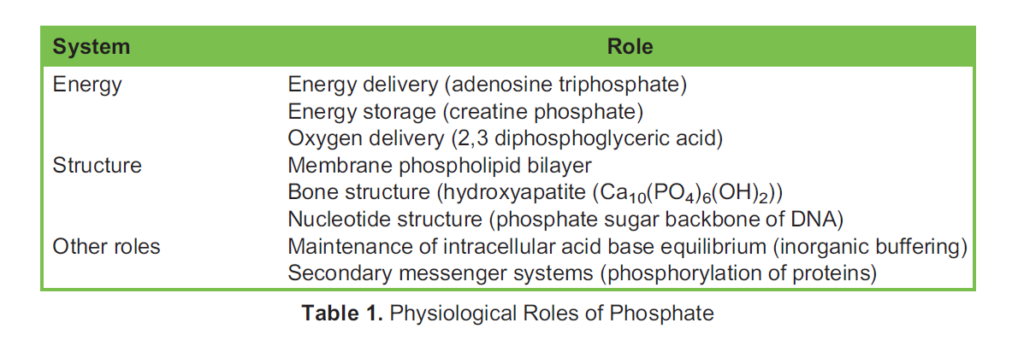
Phosphate has several key roles relevant to critical care (Table 1).3 Of note, the concentration of intracellular phosphate is considered higher than that of the plasma. It is estimated at 10 mmol/L to 75 mmol/L,5 although the true number is difficult to clarify as the total phosphate is the summation of free phosphate and numerous phosphorylated proteins that are involved in cell signalling (eg, glucose-6-phosphate, adenosine triphosphate [ATP], guanine diphosphate).
Phosphate contributes to intracellular acid-base buffering process whereby an equilibrium of dihydrogen phosphate (H2PO4–) and hydrogen phosphate (HPO42–) buffer hydrogen and hydroxide ions, respectively. Its pKa is 6.8, and intracellular pH is often less than plasma pH; thus, this is an efficient buffering system.5
Physiology of Phosphate Homeostasis
Phosphate uptake, distribution and elimination are hormonally regulated by parathyroid hormone (PTH), (1,25)-dihydroxyvitamin D3 (vitamin D) and a recently identified cohort of proteins known as phosphatonins, in particular fibroblast growth factor-23 (FGF-23).6 Phosphate homeostasis is complex and involves the gastrointestinal tract, bone, parathyroid gland and kidney (Figure 2).7,8
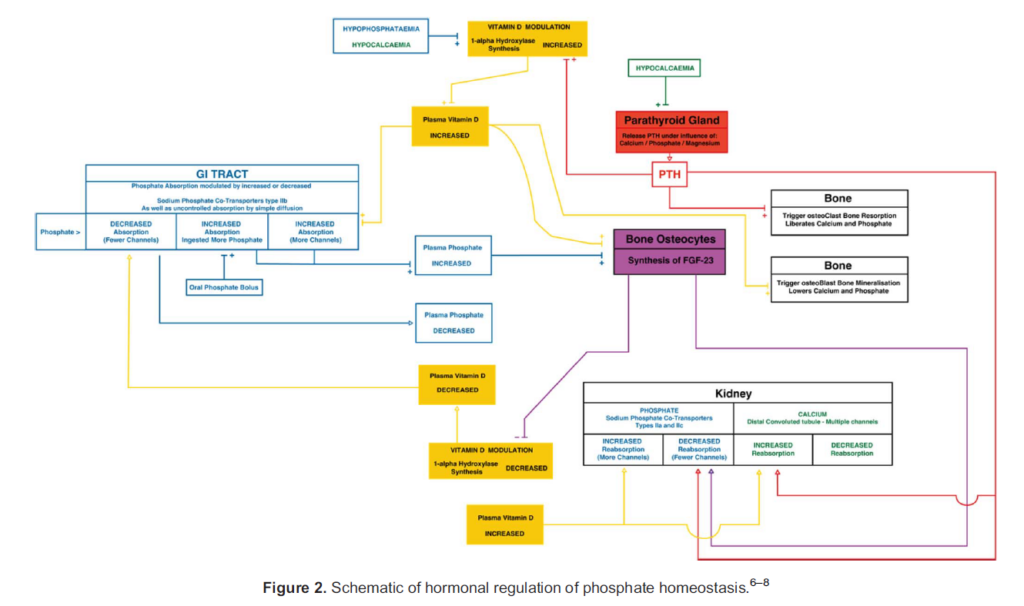
Phosphatonins
The current phosphate homeostasis model for PTH and vitamin D has been challenged by a cohort of diseases in which calcium, PTH and vitamin D levels are unaltered despite phosphate loss. Some of these diseases are tumour-induced osteomalacia (TIO), autosomal dominant hypophosphataemic rickets (ADHR) and X-linked hypophosphataemia (XLH). This observation led to a hypothesis that other humoural factors influencing phosphate homeostasis are likely to be present and were collectively described as phosphatonins.3,6 Increasing focus on the pathophysiological basis of TIO, ADHR and XLH has identified a wide range of peptides that influence phosphate homeostasis, such as FGF-23, FGF-7 and secreted frizzled related protein-4 (sFRP-4).6
An example of the role of these new factors is hyperphosphataemic tumoural calcinosis, a hereditary disorder leading to hyper- phosphataemia and deposition of excess calcium often occurring in periarticular soft tissues. This occurs due to reduced renal phosphate excretion within the proximal convoluted tubules and reduced levels of activated vitamin D. FGF-23 is implicated in this process.6 Phosphatonins and their role in disease remain an area of further research.
HYPOPHOSPHATAEMIA
Hypophosphataemia is defined as a serum concentration less than 0.8 mmol/L, which is further classified into mild (0.8- 0.6 mmol/L), moderate (0.5-0.4 mmol/L) and severe (<0.32 mmol/L). Severe hypophosphataemia is often associated with the development of overt complications,9 such as muscular weakness, impaired oxygen delivery or, in extremes, dysrhythmias and haemolysis. Hypophosphataemia in the critically ill population is often multifactorial, either due to a devel-oped deficit or a redistributive process. This redistributive hypophosphataemic state often corrects as the causative disease process resolves.
Malnourished patients who are chronically depleted of phosphate, who are subsequently inadvertently starved in the event of acute illness or surgery, have an increased risk of refeeding syndrome. This is an avoidable but life-threatening complication in which hypophosphataemia is a sign not to miss. This is elaborated upon in greater detail below.
Common Causes
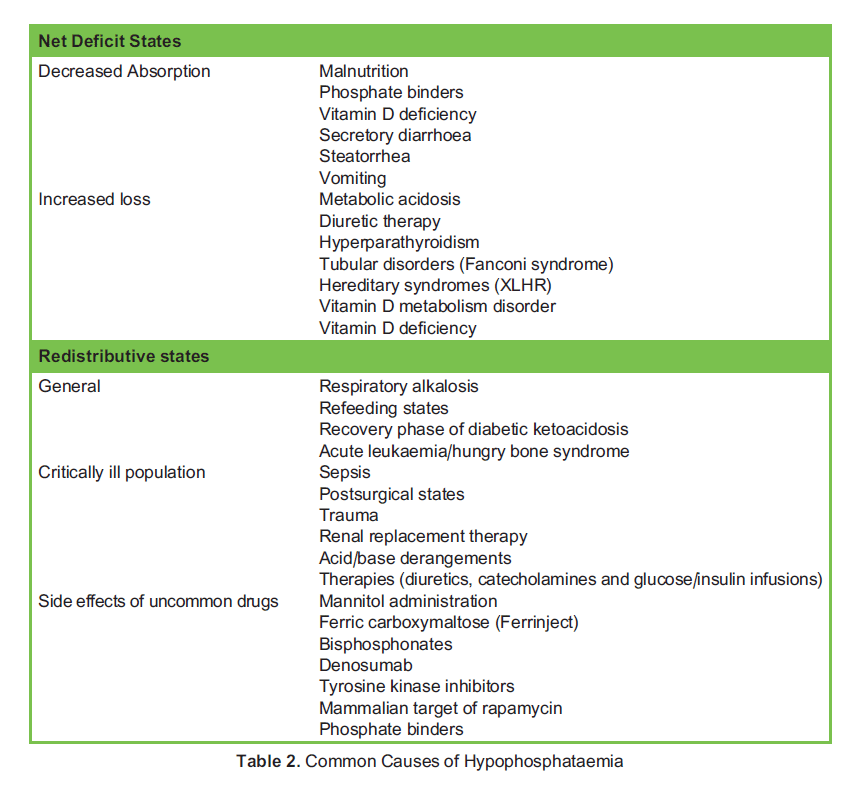
Hypophosphataemia in a patient can be approached in an ordered manner considering whether there is a deficit of phosphate overall or if there is a redistributive or sequestering process occurring (Table 2).7
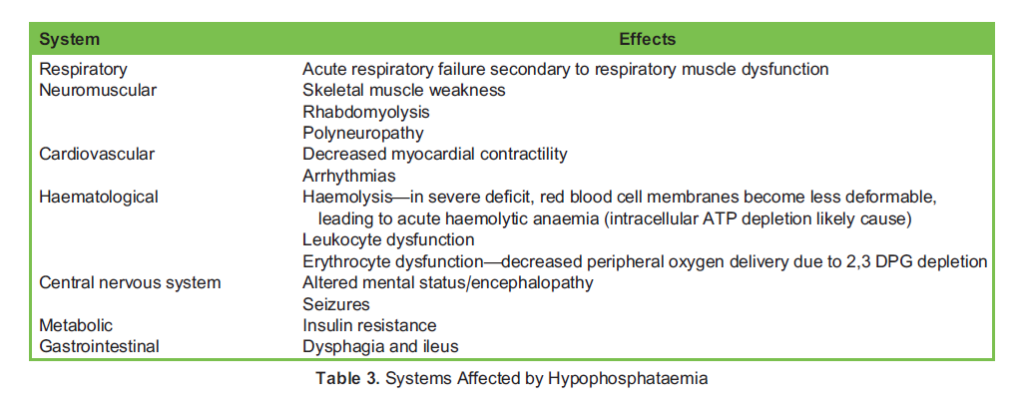
Systems Affected by Hypophosphataemia
Given the wide range of physiological roles phosphate holds, its derangement leads to dysfunction in many systems, influencing nerve and muscle conduction, including the diaphragm and cardiac muscle. The haematological system may also be affected, impairing the activity of leucocytes and, in extreme states, causing red cell haemolysis (Table 3).
Refeeding Syndrome
Refeeding syndrome is a life-threatening condition precipitated by reinitiation of feeding in a patient who has had a prolonged period of inadequate nutrition. The duration of starvation can vary, but approximately 5 days of no nutrition, or insufficient nutritional intake for 10 days, is sufficient to cause refeeding syndrome.10 Refeeding syndrome is a state characterised by electrolyte derangement and fluid shifts, which may cause cardiac and neurologic complications.
Starvation leads to a catabolic state, characterised by a low basal metabolic rate with suppressed insulin synthesis and increased glucagon synthesis. Fatty acids and amino acids are consumed for energy whilst also depleting intracellular mineral supplies.10
Upon the consumption of calories, a switch to an anabolic carbohydrate-consuming state occurs. Insulin synthesis increases in response to raised blood sugar, and a decrease in gluconeogenesis occurs secondary to decreased glucagon synthesis.10
The insulin release triggers glycogen, fat and protein synthesis, which requires phosphate, potassium and magnesium. As a progressive depletion of minerals has already occurred, stark deficits develop as the body consumes these minerals at the reestablishment of anabolism.
Typically, within 2 or 3 days of resumed calorie consumption, low phosphate, potassium, magnesium and calcium levels develop due to the intracellular consumption. This leads to deranged osmotic gradients between the intra- and extracellular compartments, and sodium retention occurs leading to hypervolaemia, oedema and the classically described anasarca state.10 The hypervolaemic state increases cardiac workload and can lead to heart failure in refeeding syndrome. Heart failure may be further compounded by thiamine deficiency following 20 days of starvation, which, given its relatively short half-life and minimal physiological storage, may lead to wet Beriberi.11
Phosphate in particular is consumed in refeeding syndrome by multiple intracellular processes that include glucose to pyruvate metabolism with phosphate intermediaries, ATP synthesis, 2,3 DPG synthesis and nucleotide synthesis. These processes lead to intracellular phosphate depletion, which ultimately impairs cellular processes that are reliant on phosphorylation for their modulation.
Cardiac arrhythmias are the most common cause of death and likely secondary to the extent of electrolytic derangements pre- sent in severe cases. These may be preceded by confusion, coma, seizures and respiratory or cardiac failure.10
Clinical Management of Hypophosphataemia
There are several different enteral and parenteral preparations for the repletion of phosphate, with the latter delivered via peripherally intravenous (IV) route at low concentration or central venous route at high concentration. The normal adult intake of phosphate is about 35 mmol/d, with typical replacement 0.2 to 0.5 mmol/kg/d (20-40 mmol/d) up to a maximum of 50 mmol in 24 hours.4,12
Check laboratory serum phosphate, calcium, urea, electrolytes, magnesium and albumin regularly (every 6 to 12 hours). If using higher phosphate concentrations and/or faster replacement rates in severe hypophosphataemia, monitoring of calcium and potassium is recommended every 4 hours, which may be facilitated with blood gas analysis within a critical care setting, enabling prompt intervention if hyperkalaemia or hypocalcaemia develop.
Reduced phosphate doses may be necessary in patients with severe renal impairment. Existing hypocalcaemia may worsen and should be corrected prior to phosphate therapy. IV phosphate replacement can cause hypotension, hypernatraemia, dehydration and iatrogenic hypocalcaemia (see the section on hyperphosphataemia below).
The optimum rate and quantity of phosphate replacement varies within the literature. It is acceptable to use clinician-guided regimens or weight-based protocols.4,12
There is a wide variety of advice regarding phosphate replacement with limited dosing advice in the British National Formulary, which advises the clinician to seek individual product literatures. In the experience of the authors, prescribing practices vary greatly given the inherent risk of inadvertent hypocalcaemia, hypotension, hypomagnaesemia and hyperphosphataemia.4 These develop rapidly with overzealous infusion rates, and as such concerns the authors sufficiently to include dosing regimen recommendations and product advice with the view to guide phosphate therapy choices in the readership (Appendix 1).
Enteral Replacement
The enteral route is often appropriate provided there is effective gastrointestinal absorption. Oral phosphate replacement is safer than IV because the latter is more likely to cause chelation (precipitation) of calcium phosphate salts and subsequent soft-tissue calcification and nephrocalcinosis. There are several oral phosphate preparations that contain a varying quantity of other electrolytes, and these should be considered in the context of the patient during therapy. Doses of up to 96 mmol phosphate orally per day in divided doses is safe for 3 to 5 days, with daily monitoring of phosphate levels.
Typical side effects of oral phosphate replacement include gastrointestinal upset, nausea and diarrhoea. It is inadvisable to coadminister phosphate supplements with calcium, magnesium or iron supplements as well as antacids, due to physical drug interactions. Phosphate replacement in patients with acute or chronic renal dysfunction and coexisting hypercalcaemia and hypernatremia may lead to phosphate nephropathy and an inadvertent increased sodium load.
Parenteral Replacement
IV preparations are reserved for those with severe hypophosphataemia (classified as <0.32 mmol/L) and/or those in critical care where the enteral route is unavailable or impaired. Preparations can be administered peripherally or centrally depending on clinical severity, fluid balance and electrolyte state. Typically, 20 to 50 mmol of phosphate in a 24-hour period is given, with appropriate monitoring as outlined above.
HYPERPHOSPHATAEMIA
Hyperphosphataemia is defined as a serum phosphate level greater than 1.5 mmol/L, with levels greater than 3 mmol/L being associated with hypocalcaemic tetany.8,9 It is primarily seen in patients with chronic renal failure and is associated with higher morbidity and mortality in this population.
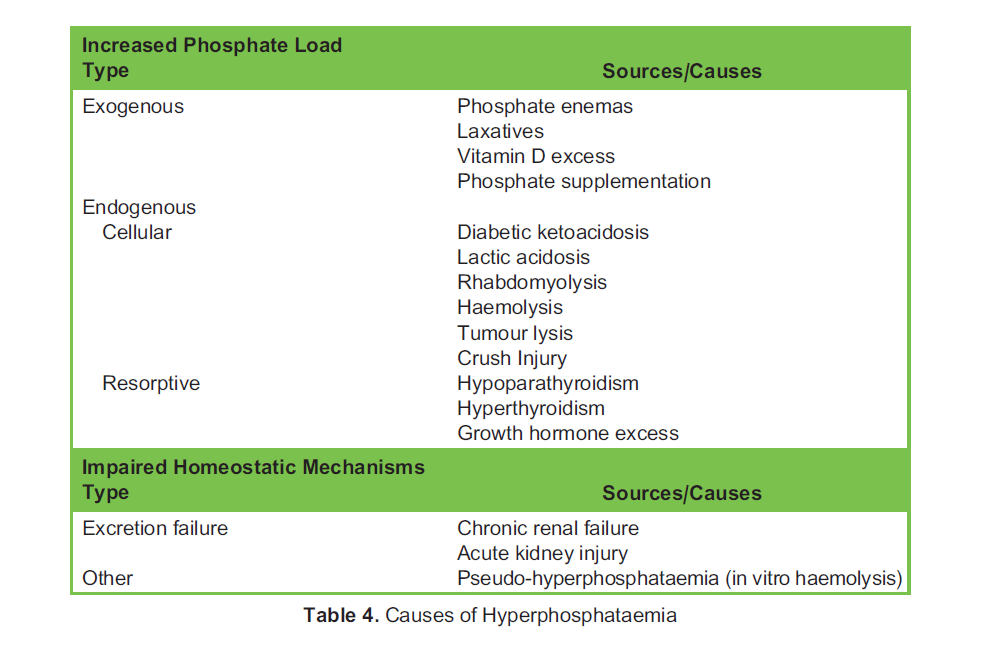
Causes
The causes of high serum phosphate levels can be organised into sources of exogenous or endogenous excess, due to redistributive processes or failure of excretion, and sometimes a combination of these processes (Table 4).8,13
Pathophysiology
In acute hyperphosphataemia, the chief consequence is low ionised calcium in the plasma due to chelation of calcium and phosphate, which precipitates in soft tissues (calcinosis) and renal tubules, causing acute phosphate nephropathy.8,13 The symptoms and signs of hypocalcaemia include paraesthesias, muscle spasms, cramping, tetany, circumoral numbness, seizures and cardiac dysrhythmias secondary to QT prolongation as well as Chvostek’s and Trousseau’s sign. If renal function is preserved, hyperphosphataemic insults such as exogenous loads are corrected in 6 to 12 hours.13
In chronic hyperphosphataemia, a secondary hyperparathyroidism state can develop, leading to increased phosphate excretion. High phosphate levels facilitate the calcification of soft tissues and vasculature such as the heart, lungs and kidneys, increasing long-term morbidity and mortality.13 Calciphylaxis, a process by which arterioles and small arteries undergo calcification, leads to a cutaneous eruption of livedo reticularis and violaceous nodules occurring on toes, fingers, ankles and but- tocks. Ulceration and haemorrhagic nodules can occur.13
There are 2 hyperphosphatemic conditions that are commonly seen in the critically ill population: tumour lysis syndrome and rhabdomyolysis.
Tumour Lysis Syndrome
Tumour lysis syndrome is caused by widespread nonapoptotic cell death, classically with haematological malignancies but also from necrosis of large solid tumours. This disordered break up of cells releases large quantities of intracellular debris
including phosphate, potassium and nucleic acids, the latter of which undergo enzymatic conversion into urate and uric acid, leading to hyperuricemia. Uric acid is poorly excreted due to its low water solubility and accumulates if the nucleic acid load overwhelms renal clearance.14
Calcium phosphate crystals form from chelation of serum calcium by released phosphate. As well, urate crystals form from hyperuricemia. Both tend to precipitate in the renal tubules, causing acute renal failure and potentially exacerbating an existing hyperkalaemia. Treatment includes managing the electrolyte disturbances, reducing the uric acid load with allopurinol and active management of uric acid levels with Rasburicase, an enzyme therapy that has urate oxidase activity. The treatment goal is to reduce the supply of substrate to the injurious process and break down uric acid product to prevent or limit renal injury, as well as manage electrolyte disturbances that are exacerbated by renal insults.14
Rhabdomyolysis
Rhabdomyolysis is the breakdown of skeletal muscle secondary to many aetiologies, including crush injury, compartment syndrome, hyperpyrexia, electrolyte disturbances and infections. It leads to a metabolic disturbance such as tumour lysis syndrome (ie, cellular debris leading to calcium phosphate precipitating in renal tubules) but with the addition of myoglobin release, which also precipitates in renal tubules causing acute renal failure. Rhabdomyolysis is classically diagnosed by measurement of creatine kinase, which peaks after the spike in myoglobin plasma concentration. Treatment, dependent on severity, involves IV or oral hydration, management of electrolyte derangement and dialysis if fluid overload, hyperkalaemia, acidaemia or symptomatic hyperuricemia develop. There is no evidence to suggest early renal replacement therapy has benefit.15
Treatment Approaches for Hyperphosphataemia
In acute hyperphosphataemia, treatment focuses on identifying and treating the cause. Phosphate excretion is facilitated by diuresis with extracellular volume expansion (ie, IV fluid therapy in excess of maintenance requirements). This may exacerbate any underlying hypocalcaemia and necessitate calcium replacement and, in the presence of renal failure, lead to volume overload. Patients should be monitored for development of signs and symptoms of volume overload.8
In situations of high phosphate with symptomatic hypocalcaemia, it may be prudent to start renal dialysis early. Intravenous calcium replacement may also be necessary to avoid life-threatening complications of severe hypocalcaemia. However, there is a theoretical risk that this may exacerbate systemic calcium phosphate precipitation, so the decision to treat needs to con- sider such risk. Often, treatment of severe hypocalcaemia would take precedence.
Phosphate binding agents are often used to reduce the dietary phosphate load, primarily to manage hyperphosphataemia in haemodialysis patients with chronic renal disease. Effective calcium-based agents include sevelamer, lanthanum and magnesium salts. Rare complications include hypercalcaemia, PTH suppression and tissue calcification.13 Recent British practice recommendations from the National Institute for Health and Care Excellence suggest that calcium-based phosphate binders are associated with poorer outcomes, based on the findings of a meta-analysis of their use.16
SUMMARY
Phosphate derangement is a common occurrence in critical care and is associated with increased disease severity and associated mortality, although whether it is causative remains to be proven. Management includes identification and anticipation of derangement, diligent replacement of deficits and treatment of any underlying pathological causes.
REFERENCES
- Suzuki S, Egi M, Schneider AG, Bellomo R, Hart GK, Hegarty C. Hypophosphatemia in critically ill patients. J Crit Care. 2013;28(4):536.e9-536.e19.
- Kim BK, Kim CY, Kim S, Kim YJ, Lee SH, Kim Associations between phosphate concentrations and hospital mortality in critically ill patients receiving mechanical ventilation. J Clin Med. 2022;11(7):1897.
- Gaasbeek A, Meinders Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005; 118(10):1094-1101.
- Geerse DA, Bindels AJ, Kuiper MA, Roos AN, Spronk PE, Schultz MJ. Treatment of hypophosphatemia in the intensive care unit: a review. Crit Care. 2010;14(4):R147.
- Hall JE, Guyton AC. Guyton and Hall Textbook of Medical Physiology. 12th ed. Philadelphia, PA: Saunders/Elsevier;
- Shaikh A, Berndt T, Kumar Regulation of phosphate homeostasis by the phosphatonins and other novel mediators. Pediatr Nephrol. 2008;23(8):1203-1210.
- Wadsworth R, Siddiqui Phosphate homeostasis in critical care. BJA Educ. 2016;16(9):305-309.
- Malberti Hyperphosphataemia: treatment options. Drugs. 2013;73(7):673-688.
- Crook M. Disorders of plasma phosphate concentration. In: Sprigings D, Chambers J, eds. Acute Medicine: A Practical Guide to the Management of Medical Emergencies. 5th ed. Chichester, UK: John Wiley & Sons; 2017:521-524. https:// wiley.com/doi/10.1002/9781119389613.ch89
- McKnight CL, Newberry C, Sarav M, Martindale R, Hurt R, Daley B. Refeeding syndrome in the critically ill: a literature review and clinician’s guide. Curr Gastroenterol Rep. 2019;21(11):58.
- Boateng AA, Sriram K, Meguid MM, Crook Refeeding syndrome: treatment considerations based on collective analysis of literature case reports. Nutrition. 2010;26(2):156-167.
- Taylor BE, Huey WY, Buchman TG, Boyle WA, Coopersmith CM. Treatment of hypophosphatemia using a protocol based on patient weight and serum phosphorus level in a surgical intensive care unit. J Am Coll Surg. 2004;198(2):198-
- Albaaj F, Hutchison AJ. Hyperphosphataemia in renal failure: causes, consequences and current management. Drugs. 2003;63(6):577-596.
- Cairo MS, Bishop Tumour lysis syndrome: new therapeutic strategies and classification: new therapeutic strategies and classification of TLS. Br J Haematol. 2004;127(1):3-11.
- Burgess Rhabdomyolysis: an evidence-based approach. J Intensive Care Soc. 2022;23(4):513-517.
- Sekercioglu N, Thabane L, Dı´az Martı´nez JP, et Comparative effectiveness of phosphate binders in patients with chronic kidney disease: a systematic review and network meta-analysis. PLoS One. 2016;11(6):e0156891.
- Phosphate Sandoz—summary of product characteristics (SmPC) – (emc). Accessed January 7, 2023. https://www.medi org.uk/emc/product/958/smpc#gref
- Kabi Summary of product characteristics potassium acid phosphate. Accessed January 7, 2023. https://www.fresenius- kabi.com/eg/documents/SPC_ADDIPHOS.pdf
- Kabi F. Summary of product characteristics sodium glycophosphate. Accessed January 7, 2023. https://www.fresenius- com/cl/documents/SPC_GLYCOPHOS.pdf
APPENDIX 1
TYPICAL REPLACEMENT REGIMENS IN THE UNITED KINGDOM17
Enteral Replacement Regimen
Enterally, up to 6 tablets per day in divided doses of Phosphate Sandoz® effervescent tablets containing sodium dihydrogen phosphate anhydrous 1.936 g, sodium bicarbonate 350 mg and potassium bicarbonate 315 mg. This is equivalent to phosphate 16.1 mmol, sodium 20.4 mmol and potassium 3.1 mmol per tablet.
PARENTERAL REPLACEMENT REGIMENS
Phosphate Polyfusor®
This preparation contains phosphate 50 mmol, sodium 81 mmol and potassium 9.5 mmol in prefilled 500-mL bottles (Table A1). Phosphate Polyfusor MUST be administered via an automated fluid pump.

Potassium Acid Phosphate (AddiPhos®)18
This preparation contains 1 mmol/mL in 10-mL ampoules and 1 mmol/mL of potassium; sodium is not present. Useful in cases of hypernatraemia/fluid overloaded states or controlled correction of hyponatraemia (Table A2).

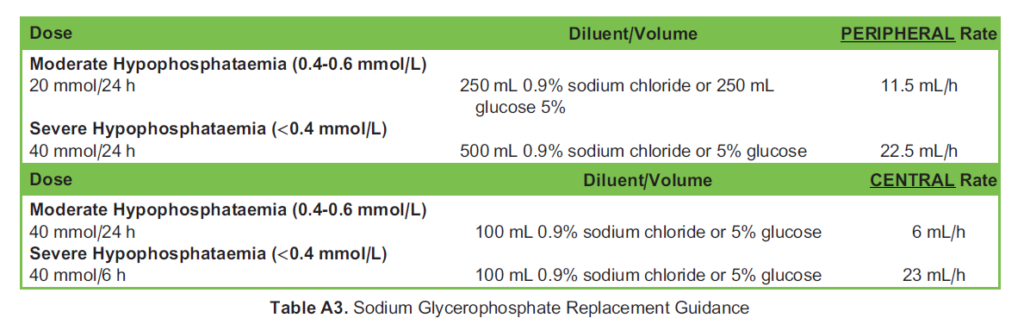
Sodium Glycerophosphate Glycophos® 21.6%19
This preparation contains 20 mmol phosphate and 40 mmol sodium in each 20-mL ampoule. Particularly useful with hyperkalaemic patients as sodium glycerophosphate contains no potassium (Table A3).
Administration Recommendations
It is advisable to administer phosphate preparations through a dedicated line as there are many incompatibilities. Phosphate Polyfusor, glycerophosphate and Addiphos preparations are incompatible with calcium- and magnesium- containing fluids, as these precipitate with phosphate in lines. In addition, Polyfusors and Addiphos are incompatible with amiodarone, caspofungin, ciprofloxacin, dobutamine and pantoprazole. A pharmacist, if available, can be helpful in guiding administration and avoiding incompatibilities.
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/