Basic Sciences

QUESTIONS
Before continuing, try to answer the following questions. The answers can be found at the end of the article, together with an explanation.
- Sevoflurane is metabolised by hepatic microsomes
- Propofol is hydrolysed by esterases.
- Chronic barbiturate therapy will induce the drug enzyme system for metabolism of halothane
- Cigarette smoking influences the choice of volatile agent for maintenance of anaesthesia.
- Sulphation is an example of a phase 1 reaction.
- Metabolism of halothane is predominately by reduction if the liver is hypoxic.
INTRODUCTION
Drugs can undergo one of several pathways on entry into the body; they can change spontaneously into other compounds e.g. atracurium breaks down into laudanosine, they can be excreted unchanged by the body e.g. excretion of benzylpenicillin by the kidneys or they can be metabolised by enzymes to different compounds to facilitate excretion e.g. ketamine. The majority of drugs follow the last route and are metabolised by enzymes within the liver; this will be the focus of the tutorial.
Most drugs are lipid-soluble and this makes them difficult to excrete. The overall aim of hepatic drug metabolism is to produce a more water soluble compound to facilitate the excretion of the drug in body fluids such as urine and bile, the primary routes of drug excretion. Very few drugs are excreted without being metabolised e.g. vancomycin.
In general, the metabolism of a drug decreases its therapeutic effect; however some drugs are metabolised firstly into active compounds before subsequent metabolism to inactive compounds. Such drugs are called prodrugs, they have no intrinsic activity before metabolism; examples include diamorphine, codeine, enalapril and levodopa. Some drug metabolites have significant activity similar to the parent compound e.g. diazepam and its metabolite desmethyldiazepam.
SITES OF METABOLISM
Many sites in the body are involved in drug metabolism including the gut wall, lungs, kidney and plasma. However, the liver is the most metabolically active tissue per unit weight and is thus responsible for the majority of drug metabolism. Other factors responsible for its contribution include its large size, it is perfused by blood containing drugs absorbed from the gut (entero-hepatic circulation) and its very high concentration of most of the drug metabolising enzymes relative to other organs. We will now look at the role of hepatic metabolism more closely.
HEPATIC METABOLISM
The smooth endoplasmic reticulum of the hepatocyte is the principal site of metabolism in the liver, with the enzymes being contained within the microsomes. The largest family of membrane-bound, non- specific, mixed-function enzymes is called the cytochrome P450 system. It contains a haem-bound iron at the active site, responsible for binding with and metabolising the drug, attached to a protein chain. It is so named because of its location (cyto = cell) and the fact that the haem moiety absorbs coloured (chrome) light at a wavelength of 450nm. The enzyme catalyses the reaction whereby a molecule of oxygen is split into its 2 atoms; one atom of oxygen is “inserted” into the substrate and one atom is reduced (combines with 2 hydrogen ions) into water, often called a mono-oxygenase reaction.
Over 50 human P450 enzymes have been identified and are classified according to their number of shared amino acid sequences. Families are given the nomenclature CYP1, CYP2 etc and are further divided into sub-families CYP1A, CYP1B etc. Sub-families can be further divided into iso-forms CYP1A1, CYP1A2 etc. Most drugs are metabolised by more than one enzyme and genetic polymorphisms do exist.
Non-cytochrome P450 enzymes in the liver are also involved in the metabolism of endogenous and exogenous compounds including, esterases and flavin-containing mono-oxygenases amongst others. Examples of commonly used anaesthetic drugs metabolised by cytochrome P450 are shown in table 1.

Table 1. Table showing examples of anaesthetic drug metabolism by cytochrome P450 system
PHASES OF METABOLISM
Metabolism of drugs is usually divided into phase 1 and phase 2. Some drugs just undergo one or the other but the majority will undergo phase 1 and then phase 2 sequentially.
Phase 1 reactions
Phase 1 reactions are classified into oxidation, reduction and hydrolysis. During these reactions, certain groups are added so that the drug can undergo the second phase to produce conjugation products. The actual activity of a drug can be altered in one of 3 ways by phase 1 metabolism; the new compound can have a similar or different activity to the parent compound, it can be converted from an active to a relatively inactive compound or from an inactive to active compound. If the metabolites of phase 1 reactions are sufficiently water soluble in nature, they can be readily excreted at this point.
Oxidation
Oxidation is the most common of the phase 1 reactions and involves the initial insertion of a single oxygen atom onto the drug molecule. Phase 1 oxidative reactions catalysed by cytochrome P450 include hydroxylation, epoxidation, dealkylation, deamination and dehalogenation reactions. The oxygen stays on the drug molecule with hydroxylation and epoxidation reactions and leaves the alkyl group as aldehyde with dealkylation reactions. Classes of drugs demonstrating oxidative metabolism include paracetamol, codeine, ropivicaine, omeprazole and phenothiazines.
Reduction
Reduction reactions are the second type of phase 1 reactions. They are also catalysed by the P450 system and often take place under anaerobic conditions. Examples of drugs subject to reduction include prednisone, a prodrug which is reduced to the active glucocorticoid prednisolone; warfarin, an anticoagulant which is inactivated by the transformation of a ketone group to a hydroxyl group and halothane which undergoes both oxidation and reduction reactions depending on the oxygen tension
present in the liver.
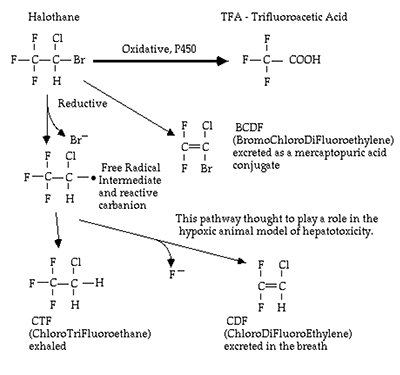
Figure 2. Diagram illustrating metabolism of Halothane
Hydrolysis
Hydrolysis is the final phase 1 reaction. Unlike the other two reactions, hydrolysis is catalysed by esterases and amidases and not by the cytochrome P450 reactions. Amide local anaesthetic agents including prilocaine undergo hepatic hydrolysis by amidases. Ester hydrolysis also occurs in extra hepatic sites; remifentanil is rapidly degraded by non-specific tissue and plasma ester hydrolysis resulting in a very short elimination half life of 3 to 10 minutes. Due to the abundance of these esterases, irrespective of the duration of an infusion of remifentanil, the time taken for the plasma concentration to halve remains unchanged. This is referred to as the context sensitive half life.
Phase 2 reactions
Phase 2 conjugation reactions involve the attachment of ionised groups to the drug and it is this process that significantly increases their water solubility, allowing excretion in the bile and urine. These reactions include glucuronidation, sulphation, acetylation and methylation amongst others. Most phase 2 reactions inactivate drugs or the active metabolites formed from phase 1 reactions.
Glucuronidation is an important metabolic pathway for many anaesthetic drugs. It is the major route of propofol metabolism, allowing excretion via the liver and kidneys (see fig 3). Morphine is metabolised to morphine-6-glucuronide which is 13 times more potent than morphine and thought to play a major role in morphine analgesia. The phase 1 metabolite of midazolam also undergoes glucuronidation. It circulates in high concentrations in the plasma and is thought to be responsible for the prolonged effects of the drug when used as sedation on intensive care units.
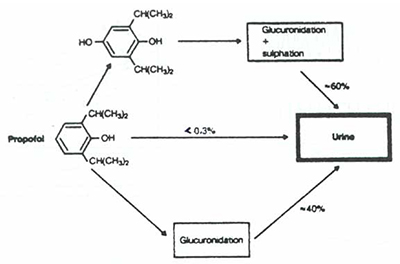
Figure 3. Diagram illustrating metabolism of Propofol
Sulphation reactions are responsible for the metabolism of about 40% of paracetamol (a further 40% undergoing glucuronidation). A small amount undergoes N-hydroxylation to N-acetyl-p-amino- benzoquinoneimine (NAPQI) which is toxic. Under normal circumstances, this is rapidly conjugated with glutathione. In paracetamol overdose this latter metabolic pathway becomes saturated and rapidly exhausts the available supply of glutathione, NAPQI accumulates and is responsible for the resultant hepatic toxicity. The treatment of a paracetamol overdose involves giving N-acetylcysteine, a precursor to glutathione.
One of the more common acetylation reactions is N-acetylation catalysed by the enzyme N-acetyl- transferase. This is important as it was the first enzyme discovered to exhibit genetic polymorphism. Individuals were classified as fast or slow acetylators depending on the variable rate of metabolism of certain drugs including isoniazid and hydralazine. Genetic polymorphism will be discussed in more detail later.
FACTORS AFFECTING DRUG METABOLISM
Various physiological and pathological factors can increase or decrease the rate of metabolism. Physiological factors include; age, sex, individual variation/genetic polymorphism, enterohepatic circulation, intestinal flora and nutrition. As a general rule, drugs are metabolised more slowly in the neonate and elderly compared with other age groups.
Pathological conditions reducing enzyme activity include aging hepatocytes in liver disease, reduced hepatic blood flow in heart failure or shock states as well as kidney disease. Drug interactions also have a large influence on the rate of metabolism by the microsomal enzymes. Drugs can be classed as enzyme inhibitors whereby they slow the rate of metabolism, or enzyme inducers whereby they speed up the rate of metabolism. It is important to know which drugs interact as depending on whether they are inhibitors or inducers, it could result in toxic or sub-therapeutic drug levels when treating patients. The effects of commonly used drugs are shown in table 2.
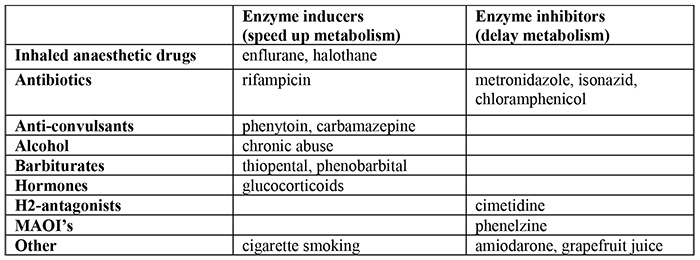
Table 2. The effects of various drugs on the microsomal enzymes
GENETIC POLYMORPHISM
Genetic polymorphism refers to the inherited differences in enzyme structure among individuals, groups or population that alters the way in which drugs are metabolised in the body. This genetic polymorphism plays an important role in anaesthesia, particulary with reference to plasma cholinesterase, acetylation enzymes and certain variants of the CYP2D6 enzymes. Plasma cholinesterase is a non-specific enzyme found in the plasma. It is responsible for the metabolism of the depolarising muscle relaxant, suxamethonium. Certain individuals have a single amino acid substitution located on chromosome 3. This results in altered enzyme activity, with significantly slower metabolism of the drug and prolonged neuromuscular blockade. There are currently 4 alleles identified; usual/normal, atypical, silent and fluoride resistant. They are distinguished by the degree of enzyme inhibition demonstrated in vitro by dibucaine, a local anaesthetic agent.
Genetic polymorphism with N-acetyltransferases involved in phase 2 metabolism produces fast and slow acetylators depending on the individual variation of the enzyme. Slow acetylators are more at risk of dose dependent drug toxicity. In the case of the cytochrome P450 system, variations can occur in up to 30% of people depending on their ethnic background
SUMMARY BOX
- Overall aim of drug metabolism is to produce a more water-soluble compound that can be excreted in urine & bile.
- Drug metabolites can have the same, increased or decreased activity compared to parent compound.
- The liver is the most metabolically active tissue
- The microsomes of the smooth endoplasmic reticulum are the site of metabolism in liver.
- The cytochrome P450 system is the predominant enzyme system. Other non-P450 enzymes are also involved however.
- Divided into Phase 1 & Phase 2 metabolism.
- Phase1 reactions include oxidation, reduction & hydrolysis.
- Phase 2 reactions are conjugation reactions.
- Drug interactions can influence the rate of drug metabolism
- Genetic polymorphisms do exist
ANSWERS
- True. Volatile anaesthetic agents are metabolised to a small extent by microsomal CYP 450 enzymes.
- False. Propofol is not an ester. It is largely glucuronidated in phase 2 metabolism.
- True. Chronic barbiturate intake induces CYP 450 enzymes, including those responsible for halothane metabolism.
- False. Cigarette smoking does not affect the choice of anaesthetic agent for maintenance of anaesthesia despite it’s affect on CYP enzymes.
- False. Phase 1 reduction reactions include oxidation, reduction, hydrolysis.
- True. Halothane undergoes both oxidation and reduction, dependent on the oxygen tension
REFERENCES
- Evan D, Karasch; MD, PhD. Principles of Drug Biotransformation. Anaesthetic Pharmacology, Physiological Principles & Clinical Practice.2004.
- R.T. Williams; Progress Report. Hepatic Metabolism of drugs. Gut, 1972, 13, 579-585.
- http://www.nottingham.ac.uk/nmp/sonet/rlos/bioproc/liverdrug/index.html
- T.E. Peck, S.A. Hill, M. Williams; Pharmacology for Anaesthesia and Intensive Care. 3rd Edition, 2008.
- http://en.wikipedia.org/wiki/Drug_metabolism
- Dr J-P van Besouw; Guide to the FRCA Examination. The Primary. September 2007.
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/