Basic Sciences

QUESTIONS
Before continuing, try to answer the following questions. The answers can be found at the end of the article, together with an explanation.
- Opioids produce analgesia by acting mainly on (single best answer):
a. Mju opioid receptor (MOP)
b. Delta opioid receptor (DOP)
c. Kappa opioid receptor
d. Orphanin/ Nociceptin receptor (NOP) - The peak analgesic effect of morphine occurs in(single best answer):
a. 1 – 2 minutes
b. 5 – 10 minutes
c. 30 – 60 minutes
d. 1 – 2 hours - Opioid receptors (True or False)
a. Are G protein coupled
b. Activate intracellular second messenger systems
c. Exhibit biased agonism
d. Are never internalised
e. Are found on immune cells
INTRODUCTION
Opioid analgesics are the gold standard in systemic analgesia for severe acute pain. There are many different compounds in clinical use around the world. Cost, regulations and clinical setting dictate their availability. There is intra-patient and inter-patient variability in response to opioids. Incomplete cross tolerance occurs when a patient is switched from one opioid to another, the clinical implication being that equivalent opioid doses need to be reduced when commencing a new opioid to avoid overdose. Knowledge of the pharmacological differences between opioids can be applied to select the appropriate drug for the relevant clinical setting and minimize the impact of side effects. Over the last 20 years more information regarding the pharmacodynamics and pharmacokinetics in terms of opioid receptor dimers and oligomers, second messenger system effects and genotyping has come to light.
Opioid analgesics exert their pharmacological actions through the μ-opioid receptor, MOP, with some also having κ-opioid receptor, KOP activity.
OPIOID RECEPTORS
Classic opioid receptors
The opioid receptor is a G-protein coupled (GPCR) with seven transmembrane regions. The opioid receptor is currently classified into:
- δ-opioid receptor, DOP (named after the tissue it was first isolated from, vas Deferens)
- κ-opioid receptor, KOP (named after the first ligand, Ketocyclazine)
- μ-opioid receptor, MOP (named after Morphine, proposed 1976,cloned 1993)
The receptors were temporarily renamed in 1996 by the International Union of Pharmacology (IUPHAR) as OP1, OP2 and OP3. Prior to this they were known as DOR, KOR and MOR. Due to the large body of literature using the Greek nomenclature, IUPHAR have recommended using the original δ,κ and μ interchangeably with DOP, KOP and MOP.
Location
These receptors are located within the central nervous system in midbrain and brain stem areas associated with descending modulatory pathways and in the dorsal horn of the spinal cord. There are also peripheral sites including the vas deferens, knee joint, gastrointestinal tract, heart and immune system.
Opioid analgesia
Activation of midbrain opioid receptors indirectly stimulates descending inhibitory pathways. These descending pathways involve serotonergic and noradrenergic transmission which results in inhibition of nocicptive traffic in the substantia gelatinosa of the dorsal horn of the spinal cord. In addition opioids can act directly on nociceptive neurons in the dorsal horn and periphery.
Nociceptin Receptor
This receptor, known as NOP was discovered in 1984. Its endogenous ligand is nociceptin/orphanin FQ. Unlike the classical opioid receptors, it does not bind naloxone, which has led to the suggestion that it should not be classified as part of the opioid receptor family. It does however have a very similar structure and intracellular mechanisms.

Table 1. Actions mediated by opioid receptors
Although, to date, only three opioid receptors have been cloned (DOP,KOP and MOP), at least 13 different opioid receptor subtypes were characterised using pharmacological methods over 10 years ago. Research is ongoing to discover the reason for this discrepancy. Postulated explanations are:
- Splice variants of the receptor (however, expression is low in the central nervous system and the intracellular C-terminal domain of the receptor is affected rather than the ligand binding extracellular N-terminal).
- Functional heterodimeric opioid receptors such as DOP-KOP and DOP-MOP may exist.
- Ligand directed GPCR signalling may produce differential effects on second messenger systems such as β-arrestin i.e biased agonism
INTRACELLULAR EVENTS
Once a ligand has bound an opioid receptor, the associated intracellular G-protein is activated. The α- subunit exchanges bound GDP for GTP and the βγ-subunit dissociates and is free to interact with intracellular second messenger systems and ion channels. With classical opioid receptor binding there is a decrease in cyclic adenosine monophosphate (cAMP) production as adenylate cylase is inhibited and also potassium conductance is increased with a reduction in calcium conductance through the cell membrane. This causes cell hyperpolarisation and reduced neuronal excitability with reduced neurotransmitter release. This is a tenable mechanism for the clinical effects of opioids but it is surprisingly unproven to date.
Other second messenger systems are coupled to activation of opioid receptors such as mitogen activated protein (MAP) kinases and the phospholipase C mediated cascade leading to the formation of inositol triphosphate and diacyl glycerol.
The concept of ligand directed G-protein coupled receptor (GPCR) signalling has recently been proposed (figure 1). GPCR activation can lead to either equal/unbiased signalling or unequal/biased signalling through G-protein and β-arrestin mediated intracellular signalling pathways. The implication is that analgesia and adverse effects may be differentially transduced by these two pathways. In β- arrestin knockout mice the anti-nociceptive effect of morphine was enhanced and prolonged whereas respiratory depression, constipation and naloxone induced withdrawals were attenuated.
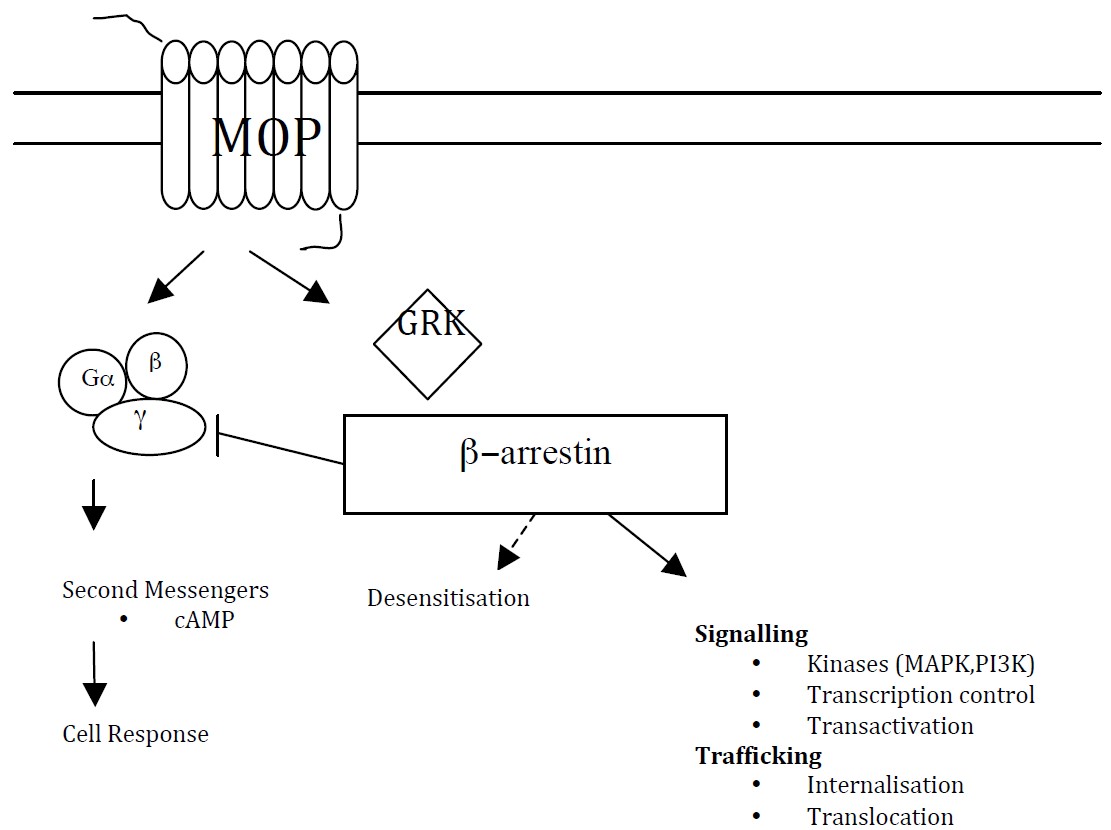
Figure 1. Opioid receptor and intracellular cascade. Mitogen activated protein kinase(MAPK). Phosphoinositide 3-kinase (PI3K) G protein-coupled receptor kinase (GRK)
LONG TERM OPIOID ADMINISTRATION
Prolonged exposure to opioids leads to multiple adaptations in second messenger signalling systems which may be responsible for tolerance, sensitisation and withdrawal symptoms.
Intracellular protein kinases are responsible for an acute phosphorylation of the MOP and DOP opioid receptors, which results in tolerance to the effects of an agonist.
Internalisation of receptors is common to all G-protein coupled receptors and is controlled by mechanisms separate from the agonist receptor interaction. G-protein coupled receptor kinases (GRK) phosphorylate agonist-bound receptors, promoting interactions with β–arrestins, which interfere with G protein coupling and promote receptor internalisation. Receptor internalisation can have divergent responses with either receptor degradation causing loss of function or receptor dephosphorylation and recycling to the cell surface with enhancing signalling.
Superactivation of adenylyl cyclase occurs with a chronic administration of opioid agonists. The alteration in levels of cAMP brings about numerous secondary changes.
Opioid induced hyperalgesia
OIH is a paradoxical response to an opioid agonist whereby instead of an analgesic, or antinociceptive effect occurring, there is an increase in pain perception.
Mechanisms of opioid induced hyperalgesia include:
- Upregulation of excitatory neurotransmitters such as substance P and CGRP in primary afferent fibres and the spinal cord.
- Increased evoked release of excitatory transmitters in the spinal cord.
- Upregualtion of spinal dynorphin levels, promoting enhanced input from afferent nociceptors.
- Activation of descending pan facilitation from the rostroventral medulla.
- Increased cholecystokinin (CCK) in the brainstem acting via descending pathways.
- TRPV1 receptor antagonists have been shown to reverse OIH.
- NMDA receptor mechanisms in keeping with central sensitization.
- Glial activation via toll like 4 receptors.
DUAL OPIOID THERAPY
Combinations of analgesics often yield pharmacological effects greater than the sum of the two. This is termed synergy. Methadone and morphine demonstrated synergy in animal models of analgesia. Interestingly the effects on gastrointestinal transit did not show synergy.
GENETICS
Splice Variants
A single gene (OPMR1) has been associated with MOP. Genes consist of exons and introns. In the normal situation the introns are spliced out so the combined exons can be transcripted into mRNA and then translated into receptor proteins. Alternative splicing (splice variants) is one way a single gene can produce a vast array of different proteins. Mice lacking exon 1 of MOP were insensitive to morphine and those lacking exon 2 were sensitive to morphine but did not have an antinociceptive response to diamorphine (heroin), fentanyl or morphine-6-glucuronide. The relevance of these finding to clinical variability is unclear at present.
Genetics come into play when metabolism of opioids is considered and codeine is an interesting case. Codeine can be considered a pro-drug and is metabolised via three pathways in the liver. The product of cytochrome P 3A4 (CYP3A4) is norcodeine, whilst codeine-6-glucuronide is produced by UGT 2B7 (over 80% of codeine metabolism). Codeine-6-glucuronide has been postulated to be responsible for the analgesic effect of codeine but the CYP2A6 pathway is widely accepted as the most important (despite being responsible for less than 5% of codeine metabolism). The product of the CYP2A6 pathway is morphine. There are several phenotypes of CYP2A6 from poor metablizers where codeine lacks efficacy to ultrarapid metabolizers where there is increased formation of morphine leading to a higher risk of toxicity. This has been highlighted with a case report of an infant of a breastfeeding mother, who died due to opioid toxicity. The mother was an ultrarapid metabolizer taking 60mg of codeine a day.
RECEPTOR DIMERISATION
Opioid receptors exist as single entities but can also exist as homodimers such as KOP-KOP or MOPMOP and heterodimers such as DOP-MOP and DOP-KOP. This dimerisation alters the pharmacological properties with the affinity for highly selective agoinsits and antagonists being reduced. Partially selective agonists and endogenous opioids have a greater affinity for these dimeric complexes. These heterodimers may explain the variability in molecular and pharmacological properties of opioid receptors.
SOME SPECIFIC OPIOIDS
Morphine
Morphine is a phenanthrene derivative which is an agonist at MOP and KOP receptors. Along with codeine it is the on the WHO essential medicines list published in March 2011. Codeine may be removed from the WHO list in the next edition subject to review. It has a bioavailability of 15-50% due to an extensive first pass metabolism.It is 20-40% protein bound predominantly to albumin and the volume of distribution (VOD) is 3.4-4.7l/kg. Degree of analgesia and plasma concentration are not clearly related. Metabolism occurs in the liver to morphine-3-glucuronide and morphine-6-glucuronide and normorphine. M-6-G has analgesic effects and M-3-G has effects on arousal. Excretion occurs predominantly in the urine as the glucuronide conjugates. 7-10% appears in the faeces as conjugates morphine. The clearance is 12-23ml/min/kg and the elimination half life is 1.7-4.5 hours. The peak analgesic effect is 30 to 60 minutes after parenteral administration due to the low lipid solubility (slowing transit to the nervous system) and the duration of effect is 3-4 hours.
Hydromorphone
Hydromorphine has similar pharmacokinetics and duration of action to morphine but is 5 times more potent with a slightly faster onset of action. Glucuronidation only occurs at position 3 making it better tolerated in patients with renal impairment due to a lack of an active metabolite.
Fentanyl, remifentanil and alfentanil
These opioids are MOP agonists which are commonly used in the perioperative period. They display pharmacokinetic differences including increased lipid solubility compared to morphine resulting in faster onset and offset. Remifentanil has a short, context insensitive half life of elimination due to metabolism by non specific tissue and plasma esterases. Remifentanil has been used to generate an experimental model of hyperalgesia.
Buprenorphine
Buprenorphine is a synthetic derivative of the alkaloid thebaine. It acts as a partial agonist at MOP receptors and dissociates slowly leading to prolonged analgesia compared with morphine. It has a high affinity for, but low intrinsic activity at KOP receptors. Due to significant first pass metabolism the sublingual route is preferred. The bioavailability is highly variable even by the intramuscular route at between 40-90%. The drug is 96% protein bound in vitro and the VOD is 3.2l/kg. Metabolism occurs in the liver by dealkylation with subsequent conjugation o glucuronide – the polar conjugates then appear to be excreted in the bile and hydrolised by bacteria in the gastrointestinal tract. Excretion occurs predominantly via the faeces as unchanged buprenorphine with the remainder excreted in the urine as conjugated buprenorphine and dealkylated derivatives. The clearance is 1l/min. The elimination half life is 5 hours.
As it is a partial agonist, buprenorphine may antagonise the effect of morphine and may precipitate withdrawal in opioid dependent patients. This only tends to occur at very high doses.
Methadone
Methadone is a synthetic opioid developed in 1942. It is a lipophylic basic drug (pKa 9.2) and exists as a racemic mixture of two enantiomers, R-methadone and S-methadone. R methadone is a potent MOP and DOP agonist. The S-methadone enantiomer is inactive as a MOP agonist but acts as an NMDA receptor antagonist. Following oral administration, time to peak plasma concentration is 2.5 to 3 hours. The oral bioavailability is high at around 85%. The VOD is high in humans at 4.2-9.2 l/kg. At physiological pH 86% of methadone is bound to plasma proteins, predominantly α1 acid glycoprotein. Unlike morphine, methadone is biotransformed in the liver rather than conjugated and at daily doses less than 55mg the majority of the metabolites are cleared in the faeces. Methadone is metabolised by the cytochrome P450 enzymes. The main enzyme responsible for the N-dethylation of methadone is CYP3A4, with lesser involvement from CYP1A2 and CYP2D6. The main product of metabolism 2- ethlidene-1,5-dimethyl-3,3-diphenylprrolidine (EDDP) is inactive. There are large interindividual variations in methadone pharmacology. Renal excretion is variable and pH dependent with excretion increasing as urine pH decreases. The elimination of methadone is biphasic. The α phase is 8-12 hours and the β elimination phase is even longer at 30- 60 hours. Despite this long elimination period the duration of analgesia is 8 – 12 hours and for pain management the daily dose is often divided into 2 or 3 administrations. There is a real risk of accumulation and toxicity with repeated doses.
Tramadol and Tapentadol
Tramadol is a partial MOP agonist with an additional serotonin and noradrenaline reuptake inhibition action and tapentadol is a MOP agonist with noradrenaline reuptake inhibition. Tramadol is metabolised to an active metabolite M1, which has greater affinity for MOP than it’s parent compound.
SUMMARY
No single mechanism adequately explains the intraindividual or interindividual variability observed with opioids. Available evidence suggests that a constellation of neurobiological, demographic, medical and patient specific factors all contribute to a determining a patient’s response to a particular opioid. Opioids remain a key component in acute pain management and in cancer pain. Opioid use in chronic non-malignant pain is limited by tolerance and hyperalgesia.
- Opioids bind to G-protein coupled receptors
- Opioid analgesia is a result of direct inhibition of peripheral and dorsal horn nociceptive neurones and activation of descending inhibitory pathways.
- Knowledge of the pharmacological differences between opioids is relevant to effective analgesia and patient safety.
ANSWERS TO QUESTIONS
- a. MOP
- c. 30 – 60 minutes
- a True, b True, c True, d False e True
REFERENCES and FURTHER READING
- Pathan H. Williams J. Basic Opioid Pharmacology: an update. British Journal of Pain. 2012 (6);1;11- 16
- Dietis N. Rowbotham D.J. Lambert D.G. Opioid Receptor subtypes: fact or artifact? British Journal of Anaesthesia; 2011;107(1);8-18
- Molinari et al. Morphine-like Opiates Selectively Antagonize Receptor Arrestin Interactions. The Journal of Biological Chemistry; 2010;285(17);12522-12535
- Peng P.W.H. Tumber P.S. Gourlay D. Review Article: Perioperative pain management of patients on methadone therapy. Regional Anaesthesia and Pain. 2005(52);5;513-523
- Felden L. et al. Comparitive clinical effects of morphine and hydromorphine: a meta-analysis. British Journal of Anaesthesia. 2011; 107(3);319-28
This work by WFSA is licensed under a Creative Commons Attribution-NonCommercial-NoDerivitives 4.0 International License. To view this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/